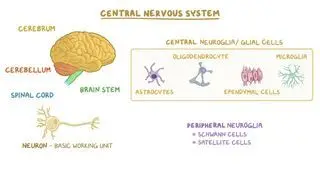
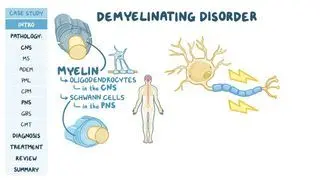
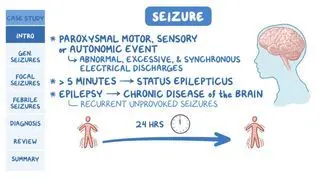

Pelizaeus-Merzbacher Disease. National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/health-information/disorders/pelizaeus-merzbacher-disease#:~:text=What%20is%20Pelizaeus%2DMerzbacher%20disease
Pelizaeus-Merzbacher Disease. Hunter’s Hope. https://www.huntershope.org/family-care/leukodystrophies/pelizaeus-merzbacher-disease/
Pelizaeus-Merzbacher Disease - Symptoms, Causes, Treatment | NORD. rarediseases.org. Accessed April 18, 2024. https://rarediseases.org/rare-diseases/pelizaeus-merzbacher-disease/#disease-overview-main
Nahhas N, Conant A, Orthmann-Murphy J, Vanderver A, Hobson G. Table 4. [Treatment of Manifestations in Individuals with Pelizaeus-Merzbacher-Like Disease 1]. www.ncbi.nlm.nih.gov. Published January 17, 2019. Accessed April 18, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470716/table/pmld1.T.treatment_of_manifestations_in_i/
Osorio MJ, Rowitch DH, Tesar P, Wernig M, Windrem MS, Goldman SA. Concise Review: Stem Cell-Based Treatment of Pelizaeus-Merzbacher Disease. STEM CELLS. 2016;35(2):311-315. doi:https://doi.org/10.1002/stem.2530
Philadelphia TCH of. Leukodystrophy. www.chop.edu. Published June 15, 2016. https://www.chop.edu/conditions-diseases/leukodystrophy#:~:text=Leukodystrophies%20are%20a%20group%20of
Garbern JY. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cellular and Molecular Life Sciences. 2006;64(1):50-65. doi:https://doi.org/10.1007/s00018-006-6182-8
Laukka JJ, Kamholz J, Bessert D, Skoff RP. Novel pathologic findings in patients with Pelizaeus-Merzbacher disease. Neuroscience Letters. 2016;627:222-232. doi:https://doi.org/10.1016/j.neulet.2016.05.028
Pelizaeus-Merzbacher Disease | National Institute of Neurological Disorders and Stroke. www.ninds.nih.gov. https://www.ninds.nih.gov/health-information/disorders/pelizaeus-merzbacher-disease