Waardenburg Syndrome · What Is It, Causes, Diagnosis, and More
Published: Mar 04, 2025
Author: Nikol Natalia Armata, MD•
Editor: Alyssa Haag•
Editor: Ian Mannarino, MD•
Editor: Kelsey LaFayette, DNP, ARNP, FNP-C
Illustrator: Jessica Reynolds, MS•
Copyeditor: Stacy Johnson, LMSW
7-day free trial
Go deeper with Osmosis
Osmosis is a learning platform with videos, questions, and AI tools to help you master topics like this.
Watch quick, visual videos
Practice with Qbank-style questions
Use AI to explain, quiz, and review
Study anytime with the mobile app
No credit card · Cancel anytime
What is Waardenburg syndrome?
Waardenburg syndrome is a rare congenital disease involving a group of genetic conditions, including distinctive facial features, discoloration of various body parts, and congenital hearing loss. The range and severity of associated findings may be highly variable, even among affected members of the same family. There are four types of Waardenburg syndrome, which differ in their characteristics and type of inheritance.
Learn deeper with Osmosis
Master this topic faster with videos, questions, and AI.
Used by 8M+ healthcare learners.
Start free trial
No credit card · Cancel anytime
What causes Waardenburg syndrome?
Waardenburg syndrome is usually caused by genetic mutations that are either inherited or occur spontaneously for unclear reasons. According to several studies, new mutations that cause Waardenburg syndrome 1 (WSI) seem to be associated with the father's advanced age. Several genes have identified mutations, like EDN3, EDNRB, MITF, PAX3, and SOX10. WSI and WSII have an autosomal dominant inheritance pattern with variable penetrance and expressivity in the affected individuals. This means that only one copy of the altered gene needs to be inherited from either parent for the child to express the disease (i.e., autosomal dominant); however, once inherited, there are a wide range of signs and symptoms that the individual may express, if at all (i.e., variable penetrance and expressivity).
On the other hand, some instances of Waardenburg syndrome type 3 (WSIII) and type four (WS4) are inherited as autosomal recessive traits, meaning two copies of the affected gene are required to be inherited from each parent for the syndrome to develop.What are the signs and symptoms of Waardenburg syndrome?
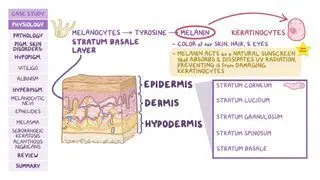
Primary features of Waardenburg syndrome include distinctive facial abnormalities; unusually diminished pigmentation (i.e., partial albinism) of the hair, skin, or irides of both eyes; and deafness present from birth.
WS type II may be distinguished from WS type I due to an absence of dystopia canthorum. Some individuals with WS present with an unusually wide nasal bridge due to abnormal lateral displacement of the inner angles of the eyes (i.e., dystopia canthorum). In addition, pigmentary abnormalities may include a white lock of hair growing above the forehead that usually disappears with age (i.e., white forelock); patchy and atypical light regions of skin (i.e., leukoderma); premature graying or whitening of the hair; and variations in the coloration of the irides (e.g., pale blue eyes) or in different regions of the same iris (i.e., heterochromia iridis). Heterochromia iridis is usually more frequent in WSII, while the presence of a white forelock and depigmented patches of skin are more common in those with WSI. Few cases have been reported in which WS has been associated with incomplete closure of the roof of the mouth (i.e., cleft palate) or the upper lip (i.e., cleft lip).Some individuals with WS have congenital hearing loss due to abnormalities of the organ of Corti, a structure within the inner ear. In most affected individuals with WS, congenital sensorineural deafness affects both ears. Rarely, hearing impairment may affect only one ear. Evidence suggests that congenital sensorineural deafness is more frequently associated with WSII than WSI.
In some cases, the features above may be associated with bilateral malformations of the arms and hands. This disorder, which has been described as a severe presentation of WSI, is sometimes referred to as WS type III (WSIII) or Klein Waardenburg syndrome. The fourth form of WS has also been described in which primary features of WS are associated with Hirschsprung disease. This form of the disorder may be referred to as WSIV, Waardenburg Shah syndrome, or Waardenburg-Hirschsprung disease. Hirschsprung disease, also known as aganglionic megacolon, is a gastrointestinal (GI) disorder of the large intestine characterized by the absence of specific nerve cells (i.e., ganglia) in the smooth muscle wall of the intestine. As a result, the involuntary, rhythmic contractions of the GI tract are affected. Associated symptoms and findings may include atypical accumulation of feces within the colon; widening of the colon above the affected segment (i.e., megacolon); abdominal bloating; vomiting; lack of appetite; failure to grow and gain weight at the expected rate, or other abnormalities.How is Waardenburg syndrome diagnosed?
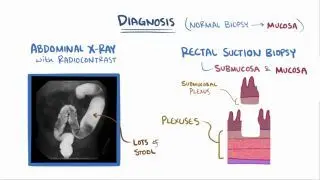
Waardenburg syndrome is usually diagnosed at birth or early childhood based upon a complete patient and family history, a thorough clinical assessment, identification of distinct physical features, and several specialized tests. More specifically, in individuals with suspected WS, the diagnostic evaluation may include using a caliper (i.e., an instrument with two movable parts used to measure thickness or diameter) to evaluate the distance between the inner angles of the eyes (i.e., inner canthi). Additional diagnostic studies may be conducted to help detect specific abnormalities potentially associated with WS, which may include examination with an illuminated microscope to visualize internal structures of the eyes (i.e., slit-lamp examination); specialized hearing tests; and advanced imaging techniques, such as to evaluate skeletal defects (e.g., seen in WSIII), or Hirschsprung disease (e.g., seen in WSIV). Computed tomography (CT) scanning may also help characterize inner ear defects responsible for congenital sensorineural deafness. Diagnostic evaluation of biopsies from specific tissue samples, such as rectal biopsies, may assist in confirming Hirschsprung disease. Lastly, genetic testing can confirm and precisely identify some genes responsible for Waardenburg syndrome. More specifically, the Waardenburg panel can detect mutations in seven genes (i.e., EDN3, EDNRB, MITF, PAX3, RET, SNAI2, SOX10).
How is Waardenburg syndrome treated?
The treatment of WS aims initially to resolve the symptoms that are apparent in each individual. Such treatment may require the coordinated medical advice of professionals, such as dermatologists, ophthalmologists, ENT clinicians, physical therapists, and other health professionals. Early recognition of sensorineural deafness may be essential in ensuring appropriate hearing aids, such as cochlear implants. This device uses electrodes implanted in the inner ear, stimulating the auditory nerve to send signals to the brain. In addition, early, special instruction may be recommended to assist in developing speech and specific methods (e.g., sign language, lip reading, communication devices, etc.) that may aid communication.
Individuals with pigmentary skin abnormalities may be prone to sunburns and have a higher risk for skin cancer. Therefore, avoiding direct sunlight, using sunscreen with a high sun protection factor (SPF), and wearing sunglasses and coverings that help to protect against the sun (e.g., hats, long sleeves, pants, etc.) are appropriate measures. For those with diminished pigmentation of the irides, specially tinted glasses or contact lenses may help reduce possible sensitivity to light.
Treatment for upper limb abnormalities may include physical therapy and potential surgical interventions. Surgery is also recommended to treat other abnormalities associated with the disorder. In individuals with Hirschsprung disease, treatment may require removal of the affected intestinal region and surgical rejoining of healthy intestinal areas.
Lastly, genetic counseling may also benefit the affected individuals and their families.What are the most important facts to know about Waardenburg syndrome?
Waardenburg syndrome includes various genetic conditions, such as distinctive facial features; unusually diminished pigmentation of the hair, skin, or eyes; and congenital hearing loss. The range and severity of associated findings may be highly variable. Waardenburg syndrome is usually caused by genetic mutations inherited from one or both parents or occurs spontaneously. Waardenburg syndrome is divided into four subtypes according to the characteristics and type of inheritance. WS type I, the presence of dystopia canthorum, as opposed to WS type II, which presents similarly but without dystopia canthorum. WS type III includes bilateral malformations of the arms and hands, and WS type IV is associated with Hirschsprung disease. Since Waardenburg syndrome can be congenital, it is usually diagnosed at birth based on a complete medical history, clinical assessment, and specialized tests. The treatment of WS aims initially to resolve the symptoms that are apparent in each individual. Genetic counseling of the parents is also strongly suggested.
Related topics

Students say Osmosis is 100% worth it
Because Osmosis saves them time. Lowers stress. And actually helps them remember when it counts.
I used Osmosis to prepare for my first medical school licensing exam! Super helpful and interactive for people who may not do great with just pages of text info!
Cecilia Ruiz
MD student

I have used Osmosis for about four years. Best thing I have ever used for my medical studies.
Sayan Misra
Med student
Osmosis videos are superior because they define simple concepts, tell a story with a clear progression, and provide context.
Jay Pate
Dental student
References
Shrinkhal, Singh A, Mittal SK, Agrawal A, Verma R, Yadav P. Waardenburg syndrome with dry eyes: A rare association. Taiwan J Ophthalmol. 2019;9(3):198-201. Published 2019 Sep 12. doi:10.4103/tjo.tjo_103_18
Sil A, Panigrahi A. Visual Dermatology: Waardenburg Syndrome Type II. J Cutan Med Surg. 2020;24(3):305. doi:10.1177/1203475420902048
Waardenburg syndrome. NORD (National Organization for Rare Disorders). (2015, June 22). Retrieved May 7, 2022, from https://rarediseases.org/rare-diseases/waardenburg-syndrome/