Amyotrophic lateral sclerosis (ALS): Nursing
Notes
| AMYOTROPHIC LATERAL SCLEROSIS (ALS) | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
Transcript
Amyotrophic lateral sclerosis or ALS, also called Lou Gehrig’s disease, is a progressive neuromuscular disease characterized by damage and degeneration of the upper motor neurons in the brain, as well as the lower motor neurons in the spinal cord.
Now, let’s quickly go over the physiology of the neuromuscular system, which includes muscles and the motor neurons serving them to ultimately trigger voluntary muscle contraction.
When we want to perform a movement, the motor cortex of the brain sends electrical impulses through an upper motor neuron. In turn, this neuron travels down the lateral corticospinal tract cord, until it reaches a second neuron, called the lower motor neuron, which relays the electrical impulses. Then, the lower motor neurons give rise to the peripheral nerves that carry the electrical impulses directly to the desired muscle fibers, which ultimately contract.
Now, the exact cause of ALS is unknown, with some evidence suggesting the cause to be excessive levels of the neurotransmitter glutamate, which can cause neurons to become overexcited, leading to damage and even death. In addition, between five to ten percent of ALS cases have a mutation in the C9ORF72 gene, suggesting that the disease can be inherited.
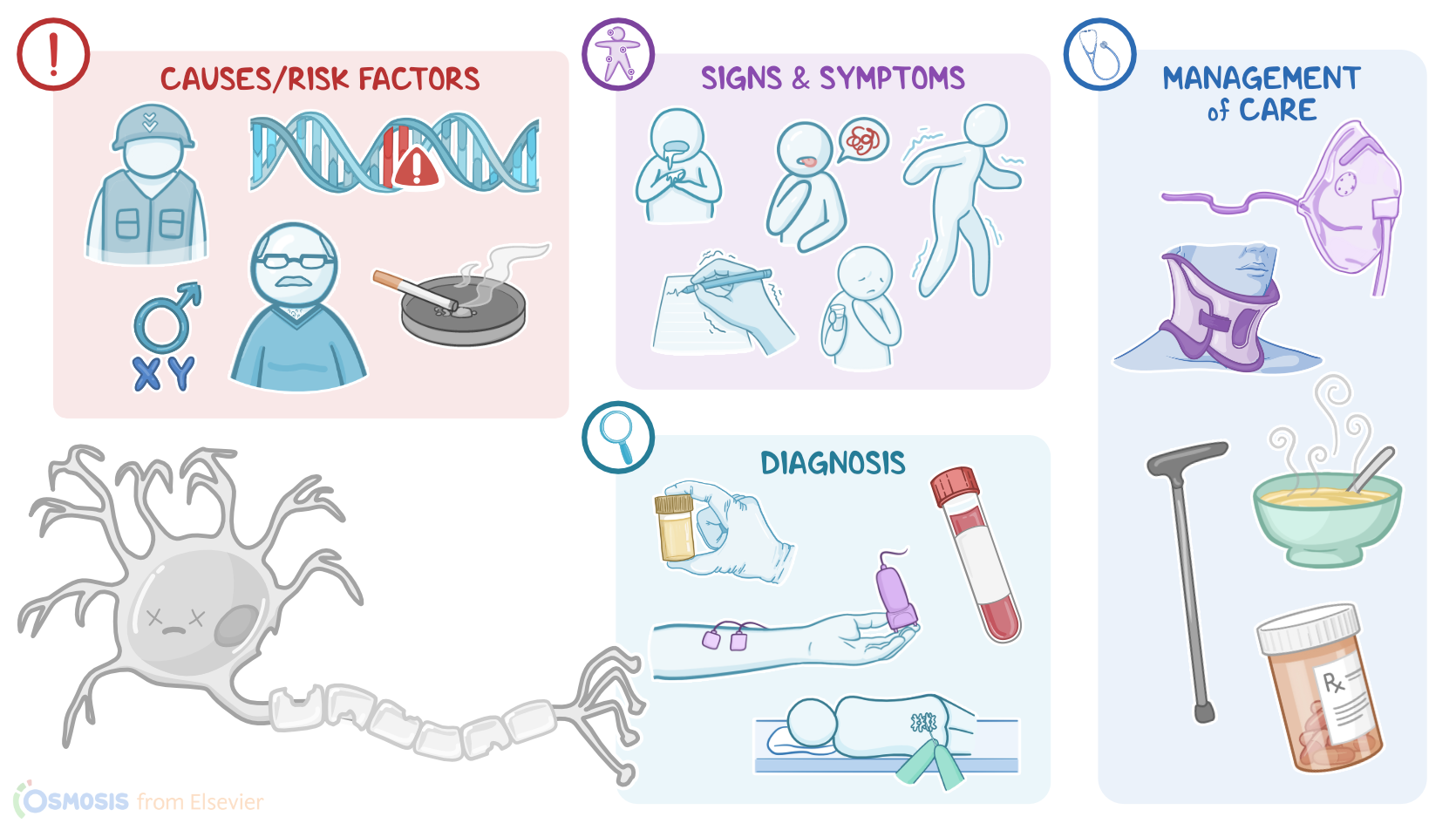
Risk factors of ALS can be grouped into modifiable and nonmodifiable risk factors. Modifiable risk factors include smoking and participating in military wars, especially in Gulf wars, where individuals are often exposed to traumatic injuries and the inhalation of chemicals and metals.
On the other hand, non-modifiable risk factors include white race, being assigned male at birth, in addition to age between 40 and 60 years, and family history.
The pathology of ALS starts with the degeneration of upper and lower motor neurons in the brain and the spinal cord respectively. This weakens the ability of neurons to transmit electrical impulses, and leads to progressive degeneration of muscles, a process called amyotrophy. As a result, the messages that originate from the motor cortex of the brain won’t reach the muscles to trigger voluntary contraction. As the motor neurons die, sclerosis occurs at the lateral columns of the spinal cord, causing them to become hardened. Now, due to its progressive nature, ALS tends to worsen overtime, until eventually reaching total muscle paralysis.
The clinical manifestations of ALS start in the limbs, and typically include progressive muscle weakness and atrophy, in addition to difficulties with fine motor movements and being easily fatigued.
Clients with ALS can also present with other motor manifestations, such as involuntary contractions, as well as spasticity or increased muscle tone and stiffness, and hyperreflexia or overactive reflexes. As the disease progresses, additional manifestations include slurred speech, difficulty swallowing, and drooling, as well as gastroesophageal reflux.
Late stages of the disease can even involve respiratory muscles, including rib cage muscles and the diaphragm. As a result, clients can develop complications like pneumonia and respiratory failure.
Additionally, due to the debilitating nature of the disease, clients can often experience emotional lability, depression, and sleep disorders. Bowel and bladder function is not typically affected by ALS. Mental capacity often remains intact, as well, though frontotemporal dementia can be present. Unfortunately, most cases of ALS lead to death within two to five years from the diagnosis.
The diagnosis of ALS starts with the client's history and physical assessment, followed by tests to rule out other causes. These tests include blood and urine tests, in addition to spinal tap to rule out inflammatory nerve conditions; as well as imaging like MRI to rule out conditions like brain tumors or multiple sclerosis.
Additional diagnostic tests include nerve conduction studies or NCS, which helps detect how well the nerves can transmit impulses to the muscles; as well as electromyography or EMG, which helps detect the abnormal electrical activity of the muscles. Lastly, diagnosis of ALS can be confirmed with a muscle biopsy that shows denervation and atrophy.
Unfortunately, there’s no cure for ALS, so treatment is typically geared at relieving symptoms, psychological support, and delaying the progression of the disease. This can be achieved by administering certain medications, such as riluzole, which reduces damage to motor neurons by decreasing glutamate levels in the brain; as well as edaravone, which is an antioxidant that destroys free radicals that may cause damage to the neurons.
Alright, now let’s take a look at the nursing care you’ll provide to your client with ALS. Your priority nursing goals are to provide supportive care and emotional support.
Sources
- "Medical-surgical nursing: Concepts for interprofessional collaborative care" Elsevier (2021)
- "Lewis’s medical-surgical nursing: Assessment and management of clinical problems" Elsevier (2020)
- "Saunders comprehensive review for the NCLEX-RN examination" Elsevier (2018)
- "Disease-modifying therapies in amyotrophic lateral sclerosis" Neuropharmacology (2020)
- "¿Por qué degeneran las motoneuronas? Actualización en la patogenia de la esclerosis lateral amiotrófica" Neurología (2019)
- "The role of mitochondria in amyotrophic lateral sclerosis" Neuroscience Letters (2019)