Phenylketonuria (PKU): Nursing
Notes
| PHENYLKETONURIA (PKU) | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
Transcript
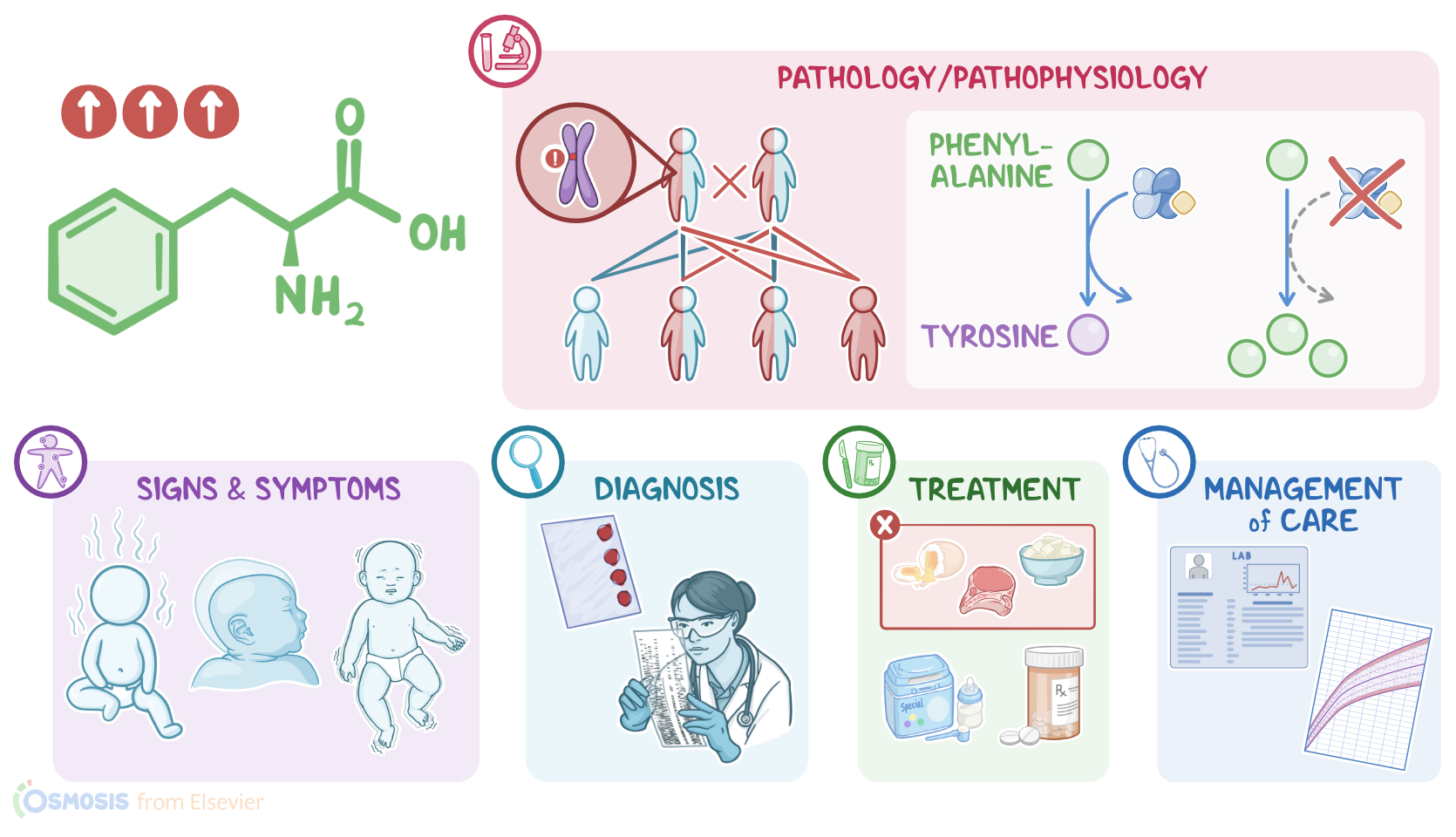
Phenylketonuria, or PKU for short, is a rare genetic disorder that causes an amino acid called phenylalanine to build up in the body. If not treated, it can damage the central nervous system, resulting in developmental delays, intellectual disability, and seizures. All right, let’s review some physiology. Amino acids are the basic building blocks that make up proteins. Phenylalanine is one of the essential amino acids, meaning our bodies can't make it so it must be acquired through protein in the diet. Since the body can’t store amino acids, any excess amino acids are converted into glucose or ketones to be used for energy. Normally, phenylalanine is converted into the amino acid tyrosine by the enzyme phenylalanine hydroxylase with the help of a cofactor called tetrahydrobiopterin, or BH4 for short. Tyrosine is then made into several other products, including dopamine and serotonin, which are neurotransmitters that neurons use to communicate; as well as norepinephrine and epinephrine, which are also neurotransmitters and hormones used by the sympathetic nervous system.
Now, PKU is an autosomal recessive genetic disorder caused by a mutation in the gene that codes for the hepatic enzyme phenylalanine hydroxylase, which helps break down phenylalanine. In autosomal recessive disorders, the client needs to inherit two copies of the mutated gene, one from each parent, to develop the condition. Because of that, PKU is more common in clients with a family history of this disorder, as well as in clients who originally come from the same region, since they frequently share versions of the same genes that have been passed down from generation to generation. Now, pathology-wise, clients with PKU have an impaired ability to use the amino acid phenylalanine due to low phenylalanine hydroxylase activity. Depending on the severity of the mutation, enzyme activity can vary from a complete absence of enzyme, resulting in very high levels of phenylalanine; to a milder form with some enzyme present but still abnormal phenylalanine levels.
Now, excess phenylalanine in the body gets converted into potentially harmful metabolites called phenylketones, such as phenylpyruvate, phenyllactate, and phenylacetate. The body has a limited ability to excrete these metabolites through the urine and sweat but they can still build up in the blood. Elevated blood phenylalanine levels can change the way the brain functions. This is because phenylalanine uses the same transporters to get across the blood-brain barrier as other amino acids, including tyrosine and tryptophan, which are essential for the formation of neurotransmitters like dopamine, norepinephrine, epinephrine, and serotonin. As phenylalanine levels rise, it occupies all the transporters, making it hard for tyrosine and tryptophan to get across the blood-brain barrier. As a result, dopamine, norepinephrine, epinephrine and serotonin levels in the brain begin to fall, and that causes problems in brain development and function.
All right, now, the clinical manifestations of PKU are absent at birth and usually appear within the first few months of life. Phenylketones in the sweat and urine can give a musty smell. Neurological symptoms can also be present, including failure to thrive, intellectual disability, microcephaly, behavioral issues, and seizures. Now, the diagnosis of PKU starts with the client’s history and physical assessment. In many countries, testing for PKU is done as part of routine newborn testing, and it detects increased levels of phenylalanine and low levels of tyrosine in the blood. The blood sample is usually taken 2 to 3 days after birth because phenylalanine levels are typically normal until the infant receives several feedings of formula or breast milk. If the screening test shows high levels of phenylalanine, the testing is repeated to confirm the diagnosis, and then genetic testing can be done to determine the exact mutation.
Treatment of PKU should begin as early as possible and must be maintained for life. It consists of a diet low in phenylalanine and high in tyrosine. This means all high protein foods, such as meat, fish, eggs, dairy, beans, and tofu, as well as some non-protein foods, such as carbonated drinks and other foods that contain the artificial sweetener aspartame, should be eliminated from the diet.Most fruit and vegetables can be eaten without limit, and small amounts of cereal and grains are often allowed. Specialized phenylalanine free formulas and medical foods are also available for infants, children, and adults with PKU. In milder cases of PKU, pharmacological treatment with a synthetic form of BH4 can be given to help make the diet less restrictive and better manage blood phenylalanine levels. Finally, clients with PKU will need to have regular blood tests to monitor serum phenylalanine and tyrosine levels.