Osmosis video - Niemann-Pick disease types A and B (NORD)
00:00 / 00:00
More Videos

Amino acid metabolism

Nitrogen and urea cycle

Citric acid cycle

Electron transport chain and oxidative phosphorylation

Gluconeogenesis

Glycogen metabolism

Glycolysis

Pentose phosphate pathway

Physiological changes during exercise

Cholesterol metabolism

Fatty acid oxidation

Fatty acid synthesis

Ketone body metabolism
Video Summary of Niemann-Pick disease types A and B (NORD)
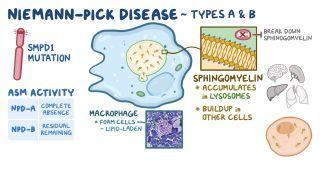
Niemann-Pick disease (NPD) type A and type B, are rare inherited conditions characterized by the inability to break down sphingomyelin, due to a deficiency of the enzyme acid sphingomyelinase. Niemann-Pick disease type A and type B result from SMPD1 gene mutation, which normally encodes to sphingomyelinase enzyme.
NPD-A symptoms present early in life and may include hepatosplenomegaly, jaundice, feeding difficulties, and progressive loss of reflexes and muscle tone. It is also often associated with a cherry red spot � in the eye, which affects the macula and impairs central vision. Usually, NPD-A becomes fatal by the age of 3 years old.
On the other hand, NPD-B represents a less severe condition that typically does not include neurologic involvement, and can develop at any time in life. Common NPD-B symptoms include progressive splenomegaly, which causes low serum platelet and white blood cell levels, high cholesterol, and declining lung function. There is no cure for NPA and NPB, and treatment is supportive and focuses on managing symptoms.