Sandhoff Disease · What It Is, Causes, Signs and Symptoms, Treatment, and More
Published: Feb 27, 2026
Author: Emily Miao, PharmD•
Editor: Alyssa Haag, MD•
Editor: Ian Mannarino MD, MBA•
Editor: Kelsey LaFayette, DNP, ARNP, FNP-C•
Editor: Mary Roberts, MSN, RN•
Editor: Anna Hernández, MD
Illustrator: Abbey Richard, MSc
7-day free trial
Go deeper with Osmosis
Osmosis is a learning platform with videos, questions, and AI tools to help you master topics like this.
Watch quick, visual videos
Practice with Qbank-style questions
Use AI to explain, quiz, and review
Study anytime with the mobile app
No credit card · Cancel anytime
What is Sandhoff disease?
Sandhoff disease, also known as Type II GM2 gangliosidiosis, is a rare inherited lysosomal storage disorder that progressively destroys nerve cells in the brain and spinal cord. Lysosomal storage disorders are caused by inherited genetic mutations that lead to defective lysosomes, which are the organelles that contain enzymes responsible for breaking down molecules like lipids, glycoproteins, and complex carbohydrates that are normally recycled within the cell. When one of these lysosomal enzymes is missing or nonfunctional, the substrate it is meant to degrade cannot be broken down and accumulates within the lysosome.
Sandhoff disease is subdivided into various forms, depending on the age of onset. The infantile form is the most common and typically the most severe form, which presents early during infancy. The juvenile form presents in early childhood and progresses more slowly than the infantile form. Finally, the adult or late-onset form presents in adolescence or adulthood and is the mildest and rarest form.
Sandhoff disease is subdivided into various forms, depending on the age of onset. The infantile form is the most common and typically the most severe form, which presents early during infancy. The juvenile form presents in early childhood and progresses more slowly than the infantile form. Finally, the adult or late-onset form presents in adolescence or adulthood and is the mildest and rarest form.
Learn deeper with Osmosis
Master this topic faster with videos, questions, and AI.
Used by 8M+ healthcare learners.
Start free trial
No credit card · Cancel anytime
What causes Sandhoff disease?
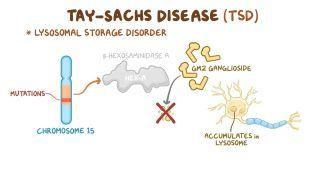
Sandhoff disease is caused by a mutation in the HEXB gene, which codes for the β-subunit shared by the lysosomal enzymes β-hexosaminidase A and β-hexosaminidase B. HEXB gene mutations cause beta-hexosaminidase A and B deficiency, which leads to an accumulation of fatty substances called GM2 gangliosides in the central nervous system and visceral organs like the liver or spleen. Gangliosides are an essential component in neuronal membranes, and they are involved in the formation of synapses during development. However, they become toxic when they can’t be degraded, leading to neuron cell death and progressive neurodegeneration.
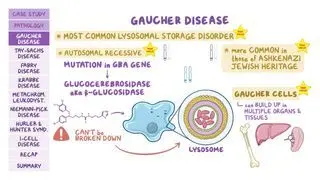
Sandhoff disease is one of the GM2 gangliosidoses and is closely related to Tay-Sachs disease, a similar lysosomal storage disorder that’s caused by mutations in the HEXA gene and affects the central nervous system. Both Sandhoff and Tay-Sachs disease are inherited in an autosomal recessive pattern, meaning two copies of the mutated gene are needed to cause the disease.
Sandhoff disease is one of the GM2 gangliosidoses and is closely related to Tay-Sachs disease, a similar lysosomal storage disorder that’s caused by mutations in the HEXA gene and affects the central nervous system. Both Sandhoff and Tay-Sachs disease are inherited in an autosomal recessive pattern, meaning two copies of the mutated gene are needed to cause the disease.
What are the signs and symptoms of Sandhoff disease?
Signs and symptoms of Sandhoff disease depend largely on how early the disorder begins. Generally speaking, the earlier the onset, the more severe and rapidly progressive the symptoms are.
Infants with the classic infantile form may not reach developmental milestones such as turning over, crawling, or holding their head up, and they may lose skills they had previously acquired. As the disease progresses, the infant may have a low muscle tone or develop spasticity and abnormal posturing. There is also often an exaggerated startle response to sound, along with seizures. Because Sandhoff disease affects not only the brain but also visceral organs, many infants develop enlargement of the liver and spleen, which can help distinguish it clinically from Tay-Sachs disease. Other symptoms include a cherry red spot in the eye and frequent respiratory infections. In most cases, the disease progresses rapidly, and most children die in early childhood, usually by 2 to 3 years of age.
The juvenile form begins later, typically in early childhood, after a period of apparently usual development. The first signs are often learning difficulties or behavioral changes, which may be mistaken initially for school or attention problems. Over time, children develop progressive cognitive decline, followed by clumsiness, coordination problems, and difficulty with speech. Seizures and vision problems are also common. Despite the slower onset, the disease is still progressive and ultimately fatal, with most individuals dying in adolescence.
Finally, adult-onset Sandhoff disease presents very differently and can be challenging to diagnose. Symptoms usually begin in late adolescence or adulthood and often progress slowly over many years. Psychiatric and behavioral symptoms are relatively common in this form and may include depression, anxiety, or psychosis, sometimes preceding neurological symptoms. Rather than severe cognitive decline, individuals commonly develop muscle weakness and wasting, sometimes resembling a motor neuron disease. Life expectancy is variable, and many individuals live well into adulthood with chronic, progressive disability.
Infants with the classic infantile form may not reach developmental milestones such as turning over, crawling, or holding their head up, and they may lose skills they had previously acquired. As the disease progresses, the infant may have a low muscle tone or develop spasticity and abnormal posturing. There is also often an exaggerated startle response to sound, along with seizures. Because Sandhoff disease affects not only the brain but also visceral organs, many infants develop enlargement of the liver and spleen, which can help distinguish it clinically from Tay-Sachs disease. Other symptoms include a cherry red spot in the eye and frequent respiratory infections. In most cases, the disease progresses rapidly, and most children die in early childhood, usually by 2 to 3 years of age.
The juvenile form begins later, typically in early childhood, after a period of apparently usual development. The first signs are often learning difficulties or behavioral changes, which may be mistaken initially for school or attention problems. Over time, children develop progressive cognitive decline, followed by clumsiness, coordination problems, and difficulty with speech. Seizures and vision problems are also common. Despite the slower onset, the disease is still progressive and ultimately fatal, with most individuals dying in adolescence.
Finally, adult-onset Sandhoff disease presents very differently and can be challenging to diagnose. Symptoms usually begin in late adolescence or adulthood and often progress slowly over many years. Psychiatric and behavioral symptoms are relatively common in this form and may include depression, anxiety, or psychosis, sometimes preceding neurological symptoms. Rather than severe cognitive decline, individuals commonly develop muscle weakness and wasting, sometimes resembling a motor neuron disease. Life expectancy is variable, and many individuals live well into adulthood with chronic, progressive disability.
How is Sandhoff disease diagnosed?
Diagnosis of Sandhoff disease begins with a thorough review of symptoms and medical history. A family history of Sandhoff, Tay-Sachs, or recurrent neonatal deaths within the family may raise suspicion of Sandhoff disease. A physical exam can identify neurologic and musculoskeletal impairments. Diagnosis is confirmed through laboratory blood enzyme testing, which detects very low or absent levels of beta-hexosaminidase enzyme activity, and genetic testing using DNA sequencing techniques, which identify a pathogenic variant in the HEXB gene.
How is Sandhoff disease treated?
There is currently no cure for Sandhoff disease, so treatment for Sandhoff disease consists of supportive care aimed at reducing symptoms. Supportive measures include adequate nutrition, hydration with fluids, respiratory therapy to prevent respiratory infections, and physical therapy to improve mobility. Anticonvulsant medications (e.g., lamotrigine, levetiracetam) can be used to treat and prevent seizures and muscle relaxants (e.g., baclofen, tizanidine) can be used for muscle spasms. There are ongoing clinical trials evaluating emerging therapies such as enzyme replacement therapy and gene therapy, however, further studies are needed to confirm their efficacy.
Individuals diagnosed with Sandhoff disease and their families can be offered genetic counseling and support groups, which can provide further education and discuss the risks of passing the disease onto future offspring.
What are the most important facts to know about Sandhoff disease?
Sandhoff disease is a rare disorder that slowly damages the nerve cells in the brain and spinal cord. It is a GM2 gangliosidosis (type II) that is closely related to Tay-Sachs disease (type I), a lysosomal storage disorder affecting the central nervous system. Sandhoff disease is caused by a mutation in the HEXB gene, which codes for enzymes beta-hexosaminidase A and B, resulting in beta-hexosaminidase A and B deficiency and a build-up of a fatty substance called GM2 ganglioside. Symptoms include progressive neurologic and musculoskeletal dysfunction, seizures, vision and hearing loss. Diagnosis is confirmed with laboratory enzyme testing, which reveals low or absent beta-hexosaminidase activity, and genetic testing, which helps identify a pathologic variant in the HEXB gene. There is currently no cure for Sandhoff disease, therefore treatment is mainly supportive care. Experimental therapies such as enzyme therapy and gene therapy are currently being studied and clinical trials are underway.
Key Takeaways
Description | Sandhoff disease is a lysosomal storage disorder that slowly destroys the nerve cells in the brain and spinal cord |
Cause | - Autosomal recessive HEXB mutation - Deficiency of hexosaminidase A and B - GM2 ganglioside accumulation |
Signs & Symptoms | - Infantile: developmental regression, seizures, hepatosplenomegaly, cherry red spot - Juvenile: cognitive decline, weakness and coordination problems, seizures - Adult: variable neurological and psychiatric symptoms |
Diagnosis | - ↓ Hexosaminidase A and B activity - Genetic testing (HEXB) |
Treatment | - No curative therapy - Supportive and palliative care - Genetic counseling |
Related topics

Students say Osmosis is 100% worth it
Because Osmosis saves them time. Lowers stress. And actually helps them remember when it counts.
I used Osmosis to prepare for my first medical school licensing exam! Super helpful and interactive for people who may not do great with just pages of text info!
Cecilia Ruiz
MD student

I have used Osmosis for about four years. Best thing I have ever used for my medical studies.
Sayan Misra
Med student
Osmosis videos are superior because they define simple concepts, tell a story with a clear progression, and provide context.
Jay Pate
Dental student
References
González-Sánchez M, Ramírez-Expósito MJ, Martínez-Martos JM. Advances in diagnosis, pathological mechanisms, clinical impact, and future therapeutic perspectives in Tay-Sachs disease. Neurol Int. 2025;17(7):98. https://doi.org/10.3390/neurolint17070098
Toro C, Tifft CJ, Zainab M. The GM2 gangliosidoses: Unlocking the mysteries of pathogenesis and treatment. Neurosci Lett. 2021;764:136195. https://doi.org/10.1016/j.neulet.2021.136195
Xiao C, Tifft CJ, Toro C. Sandhoff disease. In: Adam MP, Mirzaa GM, Pagon RA, et al, eds. GeneReviews®. University of Washington, Seattle; 2022. Accessed January 8, 2026. https://www.ncbi.nlm.nih.gov/books/