Huntington disease: Nursing
Notes
| HUNTINGTON DISEASE | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
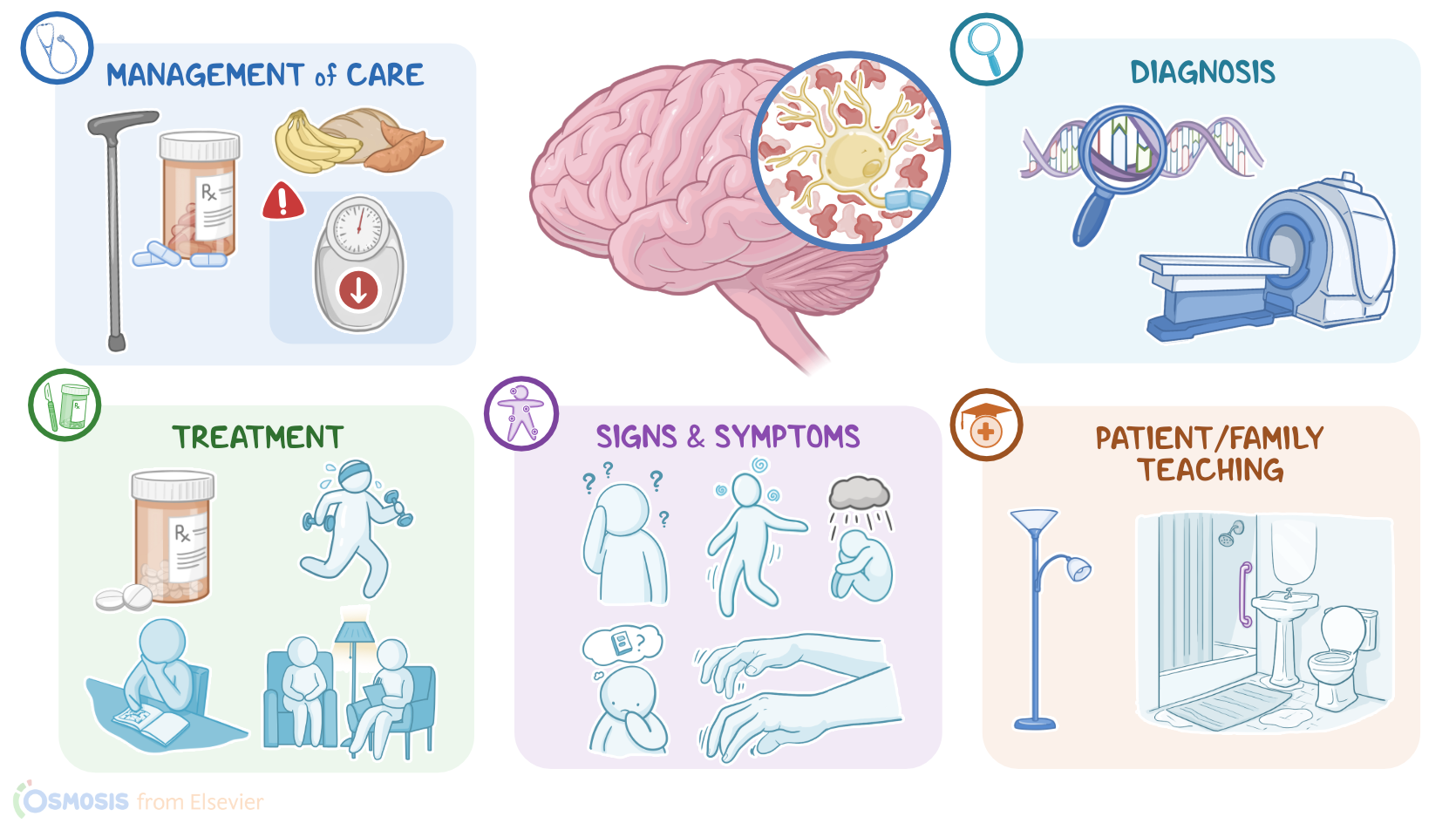
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
Transcript
Huntington disease, or HD for short, is a rare hereditary progressive neurodegenerative condition that’s characterized by motor, psychiatric, and cognitive problems. Okay, first, let’s focus on some anatomy and physiology of the brain; specifically, the basal ganglia, which are a collection of nuclei located deep in the brain. These structures help initiate, fine-tune, and complete voluntary movements. To do this, the basal ganglia receive information from the cerebral cortex, and respond by sending information to the thalamus through two pathways: the direct pathway, which is excitatory; and the indirect pathway, which is inhibitory. And there are two main neurotransmitters involved in these pathways: the excitatory neurotransmitter glutamate, and the inhibitory neurotransmitter GABA. A third neurotransmitter, called dopamine, can play both an excitatory and inhibitory role. By using the excitatory and inhibitory pathways, the basal ganglia controls what signals the thalamus sends to the motor cortex, which results in the initiation of voluntary movement.
Now, Huntington disease is caused by a mutation of the HTT gene, which is located on chromosome 4 and is responsible for the synthesis of the huntingtin protein. This mutation is inherited through the autosomal dominant pattern, meaning that one affected copy of a gene is enough to cause the disease. In other words, a person with Huntington disease has a 50% chance of passing on the affected gene to their child, so the most important risk factors are a family history of Huntington disease. The pathology of Huntington disease results from the mutated HTT gene, which codes for a defective huntingtin protein. This defective protein aggregates in the neurons of the basal ganglia, ultimately causing cell death, especially of GABAergic inhibitory neurons. As the disease progresses over time, more neurons continue to die, and eventually, the basal ganglia undergoes significant atrophy. At the same time, there are also increased levels on dopamine, and decreased levels of acetylcholine, and these neurotransmitter imbalances result in aberrant stimulation and inhibition of movement.
Clinical manifestations of Huntington disease usually occur around 40 years of age, and they include progressive motor, psychiatric, and cognitive manifestations. Motor manifestations include chorea, or involuntary, purposeless, dance-like jerking movements of the limbs and torso; athetosis, or slow, “snake-like” movements that mainly affect the hands; dystonia, which is involuntary muscle contractions that lead to repetitive jerking or twisting movement; dysphagia or trouble swallowing; dysarthria or difficulty speaking; piano playing which involves fidgeting with their hands and wrists with their arms extended; as well as abnormal eye movements, poor coordination, and unsteady gait. On the other hand, psychiatric manifestations include depression, anxiety, and paranoia; while cognitive manifestations include problems with attention, learning, poor impulse control, memory, and even dementia.
Now, there are several important complications of Huntington disease. The first one is malnutrition, since these clients may have a hard time feeding themselves due to involuntary movements and poor coordination. Another important complication is aspiration, which occurs due to discoordinated swallowing, and may result in asphyxiation. Finally, clients with Huntington disease might be at increased risk of suicidal ideation. The diagnosis of Huntington disease is based on the client’s history and physical assessment, followed by genetic testing to confirm the diagnosis. Additionally, brain imaging tests, such as CT scan or MRI, can be used to assess the atrophy of the basal ganglia. Unfortunately, there’s no cure for Huntington disease, so the treatment relies on controlling the symptoms and improving the client’s quality of life. This includes medications to control motor symptoms, such as VMAT2 inhibitors such as tetrabenazine, benzodiazepines like lorazepam, and second generation antipsychotics like risperidone; as well as medications to control psychiatric symptoms, such as selective serotonin reuptake inhibitors or SSRIs for short; Lastly, clients should try to stay active both physically and mentally, and they may benefit from psychiatric counseling and working with physical and occupational therapists.
All right, let’s look at the nursing care you will provide for your client with Huntington’s disease. Your primary nursing goals are to maintain physical safety, ensure adequate nutrition, and provide emotional and psychological support. Start by evaluating your client’s physical safety needs. Assess their ability to ambulate and perform ADLs. Institute fall precautions, and assist them when ambulating by using a gait belt and applying non-skid footwear. Collaborate with the physical therapist for the use of assistive walking devices, such as a cane or walker for added stability, as well as developing a structured rehabilitation program to promote mobility, balance, and coordination. Administer the prescribed VMAT2 inhibitor, and assist them with their ADLs such as oral care, bathing, and grooming, as needed. Next, assess their nutritional and hydration status, including skin turgor, weight, and their ability to take in oral nourishment.
Due to factors like a high caloric demand, difficulty eating related to poor coordination and dysphagia, you’ll need to collaborate with the speech-language therapist to evaluate their swallowing difficulties, as well as with the dietician to develop an individualized plan to support their nutritional needs. Offer small, frequent meals and snacks that are calorie and nutrient dense and easy to swallow, such as avocado, yogurt, mashed potatoes, eggs, and thickened liquids. During mealtimes, ensure they are in a comfortable, upright position; assist them as needed, and monitor them closely for aspiration. Report to the health care provider immediately if there’s a sudden decrease in weight or if they can no longer eat safely. If they aspirate during meals, immediately suction the airway.