Immunodeficiency disorders - Primary: Nursing
Immunodeficiency disorders - Primary: Nursing
Watch later
Watch later
Notes
| IMMUNODEFICIENCY DISORDERS - PRIMARY | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOLOGY |
| |
| CLINICAL MANIFESTATIONS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
Transcript
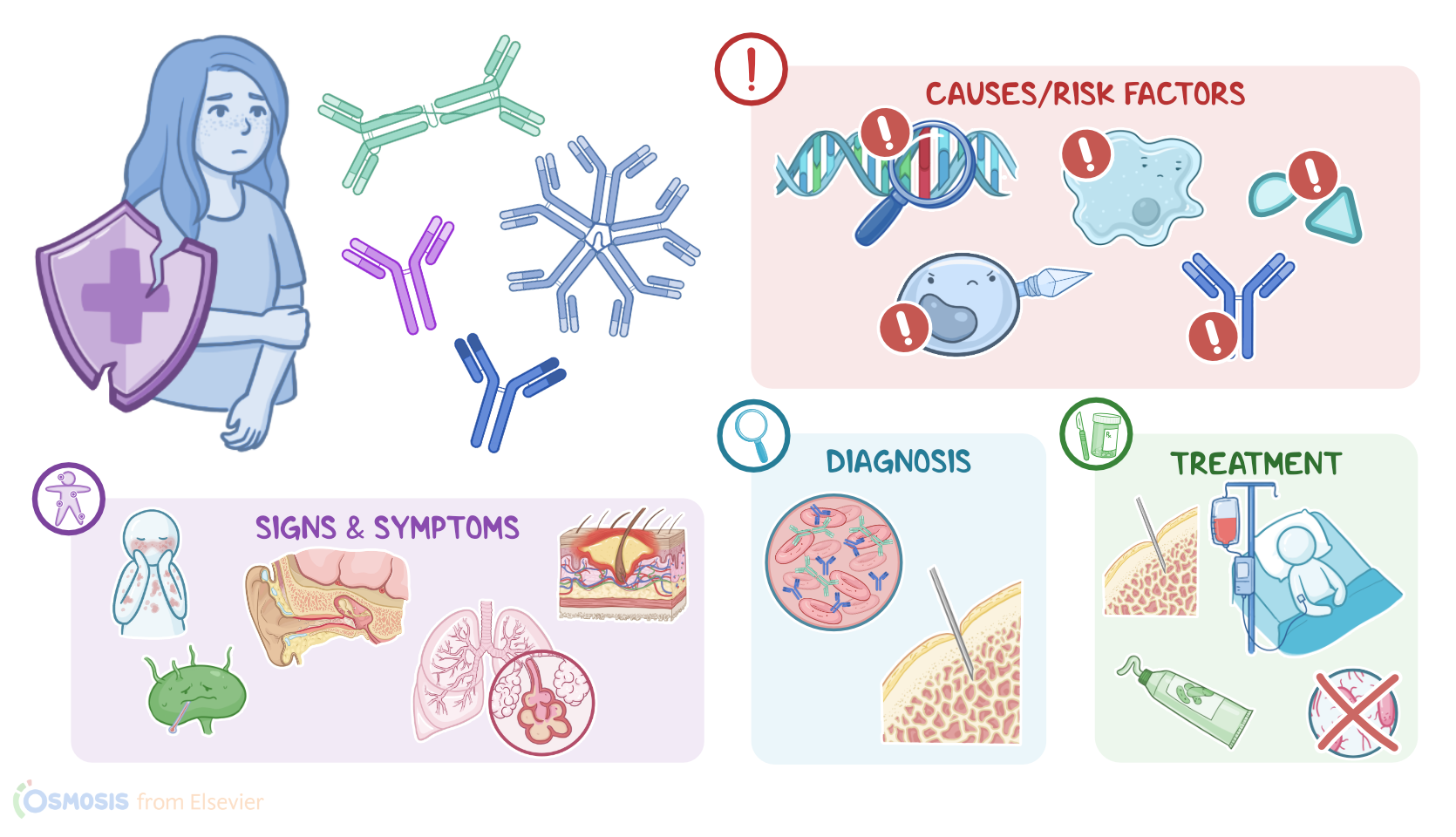
Primary immunodeficiency disorders are a diverse group of conditions that affect one or more elements of the immune system, leading to increased vulnerability to infection, autoimmune manifestations, and several types of cancer. All right, let’s quickly review some physiology. The immune system consists of white blood cells that protect us from pathogens like viruses, bacteria, and fungi, as well a foreign substances, such as toxins and chemicals, and destroy abnormal cells, such as those that might develop into cancer. Now, the immune system consists of two main branches: innate and adaptive. The innate immune response involves non-specific defense mechanisms, meaning that they do not differentiate one pathogen from another. These include complement proteins and cells like phagocytes and natural killers; as well as dendritic cells, which then activate the adaptive immune response.
The adaptive response is highly specific, meaning that it recognizes different pathogens and is mediated by cells called lymphocytes, which include T and B cells. T cells can be further divided into CD4+ and CD8+ T cells. CD4+ T cells are also known as T helper cells, because they interact with dendritic cells, and in turn help activate the rest of the lymphocytes. On the other hand, CD8+ T cells, also known as cytotoxic T cells, are in charge of cell-mediated immunity, where they attack abnormal and infected cells. Finally, B cells mediate a specific adaptive response, called humoral immunity, by secreting antibodies that bind to and destroy extracellular pathogens. These antibodies can be classified into several classes based on their structure and function, including IgM, IgA, IgD, IgG and IgE. Now, primary immunodeficiencies are a group of over 130 disorders that result from genetic defects in one or more elements of the immune system, including antibodies, T cells, complement components, and phagocytes.
Unlike secondary immunodeficiencies, which are acquired and typically occur well after infancy, primary immunodeficiencies are inherited and typically become evident during the first few years of life. So far, the only well-known risk factor for developing a primary immunodeficiency is family history. The pathology of primary immunodeficiencies varies depending on the element of the immune system that is defective. Disorders of adaptive immunity include B-cell or antibody deficiencies, T-cell disorders, and combined B and T-cell deficiencies. Since antibodies fight extracellular pathogens, antibody deficiencies are characterized by high susceptibility to pneumonia, otitis, and other infections caused by encapsulated bacteria. The most common and benign disorder within this group is selective IgA deficiency, which is characterized by low levels of IgA that normally protects mucous membranes like the digestive and respiratory tracts, with normal production of other antibody classes.
On the other end of the spectrum is X-Linked or Bruton agammaglobulinemia, a condition that almost exclusively presents in those assigned male at birth, because they have only one X chromosome. This condition results in absence of mature B cells, causing extremely low levels of circulating antibodies of all classes. Next are T-cell deficiencies; with the main one being DiGeorge or 22q11.2 deletion syndrome. This is a genetic condition where a portion of DNA on chromosome 22 is missing, causing developmental midline defects, including thymic hypoplasia. Since T-cells are produced in the bone marrow but then mature in the thymus, clients with thymic hypoplasia typically have a deficiency in mature T-cells, which results in impaired cell-mediated immunity. In turn, this results in increased risk of infection by intracellular pathogens, like viruses and fungi, as well as non-tuberculous mycobacteria. Moving on, combined B and T-cell disorders are characterized by defects in the development of both B and T cells, which respectively lead to impaired antibody and cellular immune responses.
With severe combined immunodeficiency, or SCID for short, the immune system is so dysfunctional that it’s considered almost completely absent. Clients with SCID have an extreme susceptibility to all kinds of bacterial, viral, and fungal infections; including opportunistic infections that a healthy immune system would typically be able to fend off. Finally, there are disorders of innate immunity, including phagocyte disorders and complement disorders. Phagocyte disorders are characterized by recurrent bacterial and fungal infections of the skin and mucosal membranes, impaired wound healing, and delayed separation of the umbilical cord. On the other hand, complement deficiencies involve one or more complement proteins. Early complement deficiencies involve C1 to C4, and are associated with an increased risk of severe, recurrent pyogenic infections of the sinuses and respiratory tract, as well as autoimmune manifestations like systemic lupus erythematosus.
Terminal complement deficiencies, on the other hand, involve C5 to C9, and individuals are particularly susceptible to recurrent Neisseria infections.Now, although the clinical manifestations of primary immunodeficiencies are highly variable, most disorders lead to increased susceptibility to infection, as well as a higher risk of developing autoimmune disorders, and certain kinds of cancer, especially leukemia or lymphoma. Primary immunodeficiencies should be suspected in clients with recurrent sinus or ear infections, frequent pneumonias, failure to thrive, poor response to prolonged antibiotic therapy, and a persistent thrush or skin abscesses.
So, the diagnosis of primary immunodeficiency disorders starts with the client’s history and physical assessment. Further diagnostic tests may include obtaining a complete blood count, measuring the levels of circulating antibodies, and performing a flow cytometry assay to determine the number and percentage of lymphocytes. If the diagnosis is still unclear, a bone marrow biopsy can be performed to look at the number and morphology of hematopoietic stem cells. Finally, diagnosis of certain conditions can also be confirmed with genetic testing, such as fluorescence in situ hybridization or FISH for short, and PCR, to look for the exact genetic abnormality causing the disease. Treatment of primary immunodeficiencies is aimed at preventing severe infections, treating infections with appropriate antimicrobials, as well as strengthening the immune system, and treating the underlying cause of the immune defects, when possible.