Polycystic kidney disease (PKD): Nursing
Polycystic kidney disease (PKD): Nursing
Renal/Urinary
Renal/Urinary
Notes
| POLYCYSTIC KIDNEY DISEASE (PKD) | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
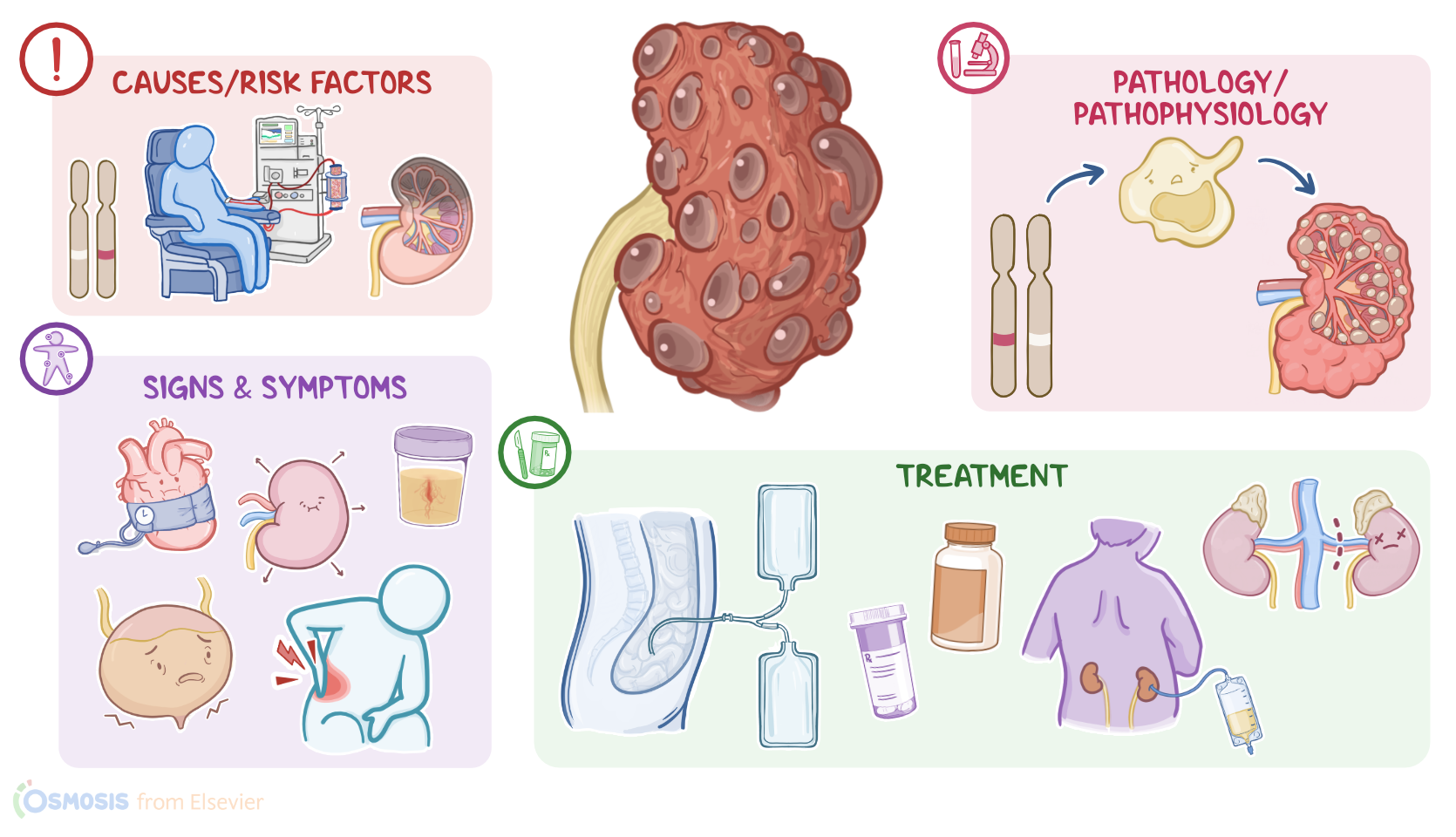
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
Transcript
With polycystic kidney disease, poly- means multiple and cystic refers to fluid-filled sacs. So, polycystic kidney disease describes a condition in which fluid-filled sacs form in the kidney. There are three types of polycystic kidney disease: infantile polycystic disease, which appears in infancy or childhood; adult polycystic disease, which appears in adulthood; these first two conditions are inherited. The third type is not inherited, and is therefore called acquired polycystic disease.
First, let’s quickly review the anatomy and physiology of the kidneys. These organs are made up of an outer cortex and an inner medulla. The cortical tissue extends towards the medulla, forming renal columns that divide the medulla into pyramidal-shaped structures called the renal pyramids. Now, the cortex and the medulla house the functional units of the kidney, called the nephrons, which filter the blood and create urine.
This urine drains from the tips of renal pyramids to the minor calyces, which then drain into the major calyces. The major calyces then merge to form the renal pelvis, which drains urine into the ureters. The two ureters carry urine to the urinary bladder, which is a pelvic organ that stores urine.
During urination, urine passes from the bladder to the urethra and to the outside of the body. The kidneys can also produce hormones, such as renin and erythropoietin. Renin raises blood pressure when it falls below normal, while erythropoietin stimulates red blood cell production from the bone marrow.
Now, the causes of polycystic kidney disease depends on the type of disease. Infantile polycystic disease is caused by a mutation of the polycystic kidney and hepatic disease gene, or PKHD1 gene for short. This type is inherited in an autosomal recessive pattern, meaning that the individual needs to receive one gene mutation from each parent to have the disease, so it’s also known as autosomal recessive polycystic kidney disease, or ARPKD.
On the other hand, adult polycystic kidney disease is caused by a mutation in the polycystic kidney disease 1 gene or the polycystic kidney disease 2 gene, called PKD1 and PKD2 genes. In contrast to the infantile type, adult polycystic disease is inherited in an autosomal dominant pattern, meaning that the individual needs to receive only one gene mutation from one of the parents to have the disease, so it’s also called autosomal dominant polycystic kidney disease or ADPKD. Finally, acquired polycystic disease is typically caused by chronic kidney disease and long-term dialysis related to kidney failure.
Alright, now the pathology of infantile and adult polycystic kidney diseases starts with PKHD gene mutation, which causes loss of cellular regulation and uncontrolled cell division. This leads to the formation of cysts in the nephrons, and these cysts enlarge over time by collecting fluid.
As cysts grow, they cause kidney damage by putting pressure on the nearby healthy kidney tissue. These cysts can also put pressure on the nearby blood vessels, lowering their blood pressure. This drop in blood pressure triggers the release of renin, which increases the systemic blood pressure as an attempt to increase the blood flow to the kidneys. In addition, growing cysts can cause enlargement of the kidney with less healthy tissue.
The result is chronic kidney disease and impairs renal functions like fluid and electrolyte balance. One of these is salt wasting where the kidneys can not retain sodium, leading to hyponatremia. The client will also have impaired urine production and waste elimination. When it comes to acquired polycystic disease, the exact mechanism is unknown, but it seems to be associated with the chronic uremia caused by chronic kidney disease. These new cysts can damage the kidneys even further, leading to additional loss of renal function.
Now, the main complications of infantile and adult polycystic kidney diseases are renal failure and hypertension, which can also manifest as preeclampsia during pregnancy. Other complications include hematuria, infections, and kidney stones that could cause urinary tract obstruction.
In some clients, cysts can also develop in the liver, blood vessels, or other organs causing cystic liver disease; cerebral aneurysms that can rupture; heart valve disorders; in addition to diverticulosis in the colon. These cysts can also enlarge, putting pressure on the lungs, which can impair breathing. Finally, for acquired cystic disease, the complications typically include cyst infection and rupture.
The clinical manifestations of polycystic kidney disease differ for each type. Signs and symptoms of infantile polycystic kidney disease can start during intrauterine life, and these include low levels of amniotic fluid, called oligohydramnios; infants that are small for gestational age; enlarged kidneys; portal hypertension, and breathing difficulties. On the other hand, clients with adult polycystic disease typically present with flank pain, hypertension, kidney stones, and urinary tract infections, in addition to hematuria, nocturia, and inability to concentrate urine. Clients with acquired polycystic disease are usually asymptomatic, but some present with fever, back pain, or hematuria. With all types, the kidneys are typically enlarged on physical assessment.
The diagnosis of polycystic kidney disease starts with the client’s history and physical assessment. Next, an abdominal ultrasound is the main method used for diagnosis. This can be followed by CT or MRI for confirmation. Imaging tests will show enlarged kidneys with multiple cysts. Lab work includes a CBC, blood urea nitrogen, or BUN for short, and creatinine levels, and the latter can be used to calculate the estimated glomerular filtration rate, or eGFR for short. Increased BUN and creatinine levels and a decrease in eGFR typically signify disease progression. Finally, urinalysis can show hematuria; proteinuria; as well as pyuria, or increased WBCs in the urine, when there’s a urinary tract infection. Serum inflammatory markers, like CRP, along with serum WBCs can also be increased with a systemic infection.
Treatment of polycystic kidney disease aims to manage symptoms with medications. These medications include tolvaptan, which increases blood flow to the kidneys to maintain their function; anti-hypertensive medications, such as angiotensin-converting enzyme inhibitors or ACEI for short; in addition to calcium channel blockers and beta blockers. Opioids and acetaminophen can also be prescribed to reduce pain, while avoiding NSAIDs and aspirin that can further impair kidney function. Other medications include potassium citrate for kidney stones and antibiotics or percutaneous drainage of the cyst in case of infected cysts.