
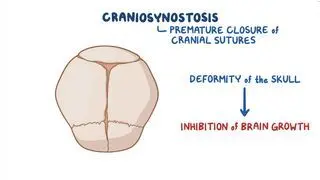


Kim HJ, Roh HG, Lee IW. Craniosynostosis: Updates in radiologic diagnosis. J Korean Neurosurg Soc. 2016;59(3):219-226. doi:10.3340/jkns.2016.59.3.219
Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995;11(4):462-464. doi:10.1038/ng1295-462
Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 1994;8(1):98-103. doi:10.1038/ng0994-98
Warren SM, Proctor MR, Bartlett SP, et al. Parameters of care for craniosynostosis: Craniofacial and neurologic surgery perspectives. Plast Reconstr Surg. 2012;129(3):731-737. doi:10.1097/PRS.0b013e3182412a50