Sickle cell disease: Nursing process (ADPIE)
Notes
| SICKLE CELL ANEMIA | ||
| KEY POINTS | NOTES | |
| PATIENT REPORT |
| |
| PATHOPHYSIOLOGY |
| |
| DIAGNOSIS AND TREATMENT |
| |
| ASSESSMENT |
| |
| NURSING DIAGNOSES |
| |
| PLANNING |
| |
| IMPLEMENTATION |
| |
| EVALUATION |
| |
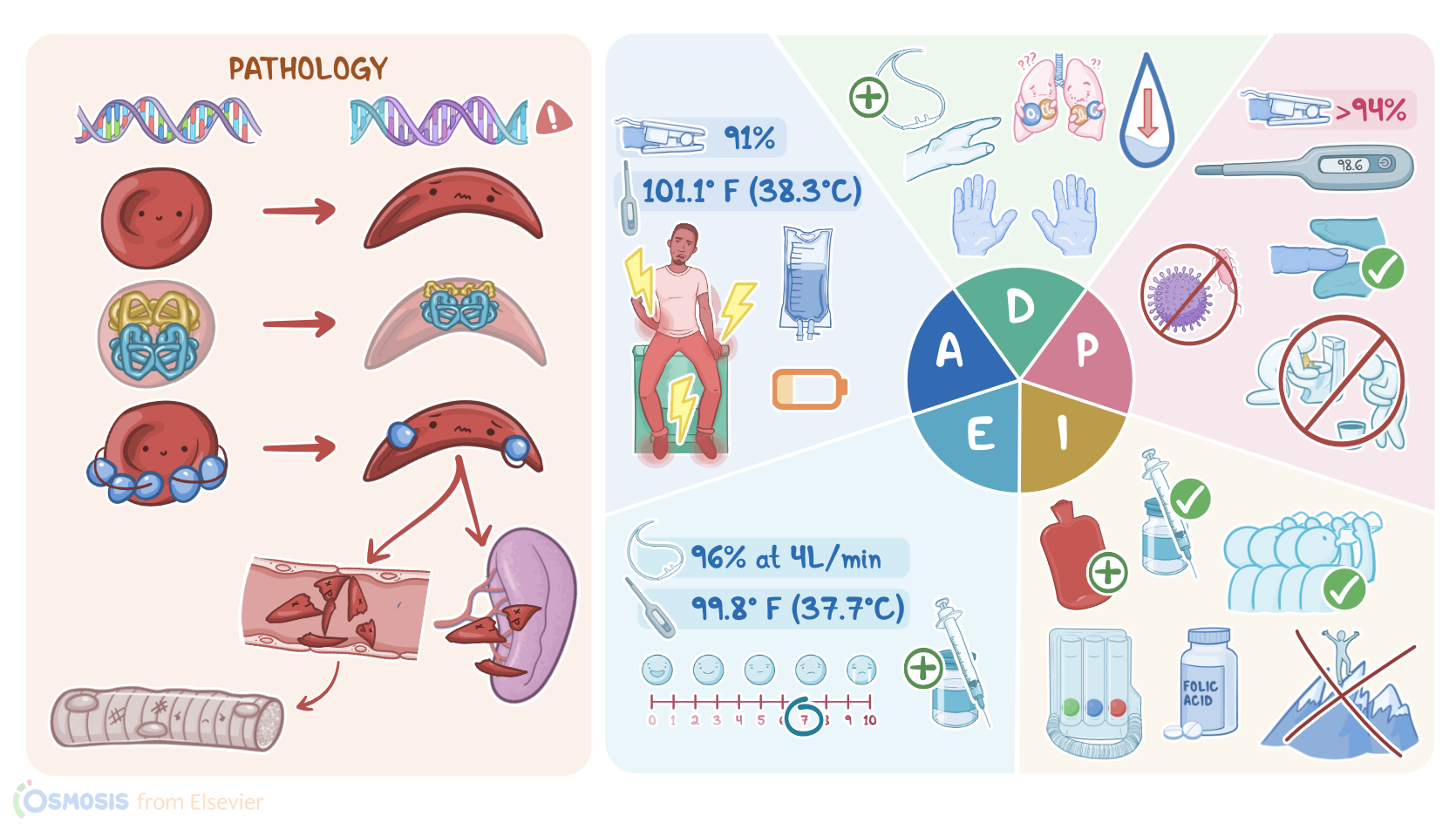
Transcript
Darnell Tyler is a 24-year-old Black male client with a history of sickle cell disease who presents to the emergency department, or ED, with a report of sudden, severe pain, rated a 9/10 that started late last night in his back, hands and feet.
He says he has been nauseous and vomiting for the last few days. After blood is drawn for culture, IV fluids and antibiotics are started. Then Darnell is admitted to the medical unit for further treatment and evaluation.
Sickle cell disease is a group of genetic conditions that affect hemoglobin, which can cause red blood cells to take the shape of a sickle or crescent. As a result, these red blood cells are more fragile and can be destroyed more easily, which can cause anemia.
Normally, red blood cells are able to carry oxygen from the lungs to peripheral tissues because they contain hemoglobin A, or HbA for short. Now, hemoglobin A is made up of two α-globin and two β-globin peptide chains, giving red blood cells a characteristic flexible biconcave shape that allows them to travel easily through blood vessels.
With this in mind, sickle cell disease is caused by a mutation in the HBB gene, which almost always results in the 6th amino acid of beta globin being a valine instead of glutamic acid.
As a result, two normal α-globin and two mutated β-globin peptide chains give rise to an abnormal hemoglobin called hemoglobin S for sickle, or HbS for short.
Now, under normal conditions, HbS is also able to carry oxygen quite well. However, under conditions such as hypoxia, acidosis, or dehydration, HbS forms long chains within the red blood cells.
This ultimately distorts the red blood cells into a rigid and fragile crescent shape that looks like a sickle. Now, sickle cell disease is autosomal recessive, so both parents must pass the mutated HBB gene to their child, so they will be homozygous for HbS.
This is especially common in individuals whose ancestors came from Sub-Saharan Africa, as well as South America, the Caribbean, and Central America. The mutated gene is also quite common among individuals of Mediterranean, Middle Eastern, Indian, and Asian descent.
On the other hand, if a client has one mutated HBB gene and one normal HBB gene, then they’re heterozygous for HbS, which makes them sickle cell carriers, also called sickle trait.
These clients usually have no health problems unless they are exposed to extreme conditions like dehydration or high altitude.Now, some of these fragile sickle cells get prematurely destroyed in blood vessels, a process often referred to as intravascular hemolysis; while others get destroyed by the spleen, which is also known as extravascular hemolysis.
Increased red blood cell breakdown leads to anemia and symptoms such as fatigue, pallor, shortness of breath, and jaundice. Less red blood cells stimulate bone marrow erythropoiesis, leading to new bone formation, and extramedullary hematopoiesis leading to hepatomegaly.
Finally, sickle cells can't travel easily through blood vessels, so they may get stuck and clump together in smaller blood vessels, causing vaso-occlusion, meaning they block the blood flow, eventually leading to tissue ischemia and pain, as well as infarction and necrosis.
This is especially common in the long bones and the back, as well as hands, and feet. So these clients typically present with bone or joint pain without a history of trauma, as well as swelling and pain in the fingers and toes.
Vaso-occlusion can result in many complications, which is known as a vaso-occlusive or sickle cell crisis. An important complication is avascular necrosis of the hips, which can cause tiny breaks in the bone and eventually, the bone may collapse.
In the brain, vaso-occlusion can lead to headaches, seizures, and even ischemic stroke. Vaso-occlusion in the lungs and cause a pulmonary infarction, which is often referred to as acute chest syndrome, and may present with chest pain, dyspnea, and cough.
Another important complication in the lungs is pulmonary hypertension, which can even lead to cor pulmonale, often referred to as right-side heart failure. In the kidneys, vaso-occlusion can cause sickle cell nephropathy, which is associated with hematuria and proteinuria; as well as chronic kidney disease.
Next, red blood cells can clog up the spleen and cause splenic infarcts; and since the spleen plays an important role in immunity against encapsulated bacteria, a client with sickle cell disease will be susceptible to infections by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae.
Finally, clogging of small blood vessels of the retina can lead to retinopathy and vision loss. To prevent these complications, it’s important to diagnose sickle cell disease as early as possible.
Diagnosis of sickle cell disease can be done prenatally with DNA testing, as well as with routine newborn screening. Hemoglobin electrophoresis can be performed to identify HbS.
Another important diagnostic study is a peripheral blood smear, which shows the characteristic sickle cells. Additionally, since there’s hemolysis, the blood levels of unconjugated bilirubin are often increased, while the hemoglobin levels are decreased.
To compensate, the bone marrow revs up and starts pumping out immature red blood cells called reticulocytes, therefore the reticulocyte count is usually elevated. In clients with bacterial infection, white blood cell count can also rise.