Cystic fibrosis: Nursing
Notes
| CYSTIC FIBROSIS | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
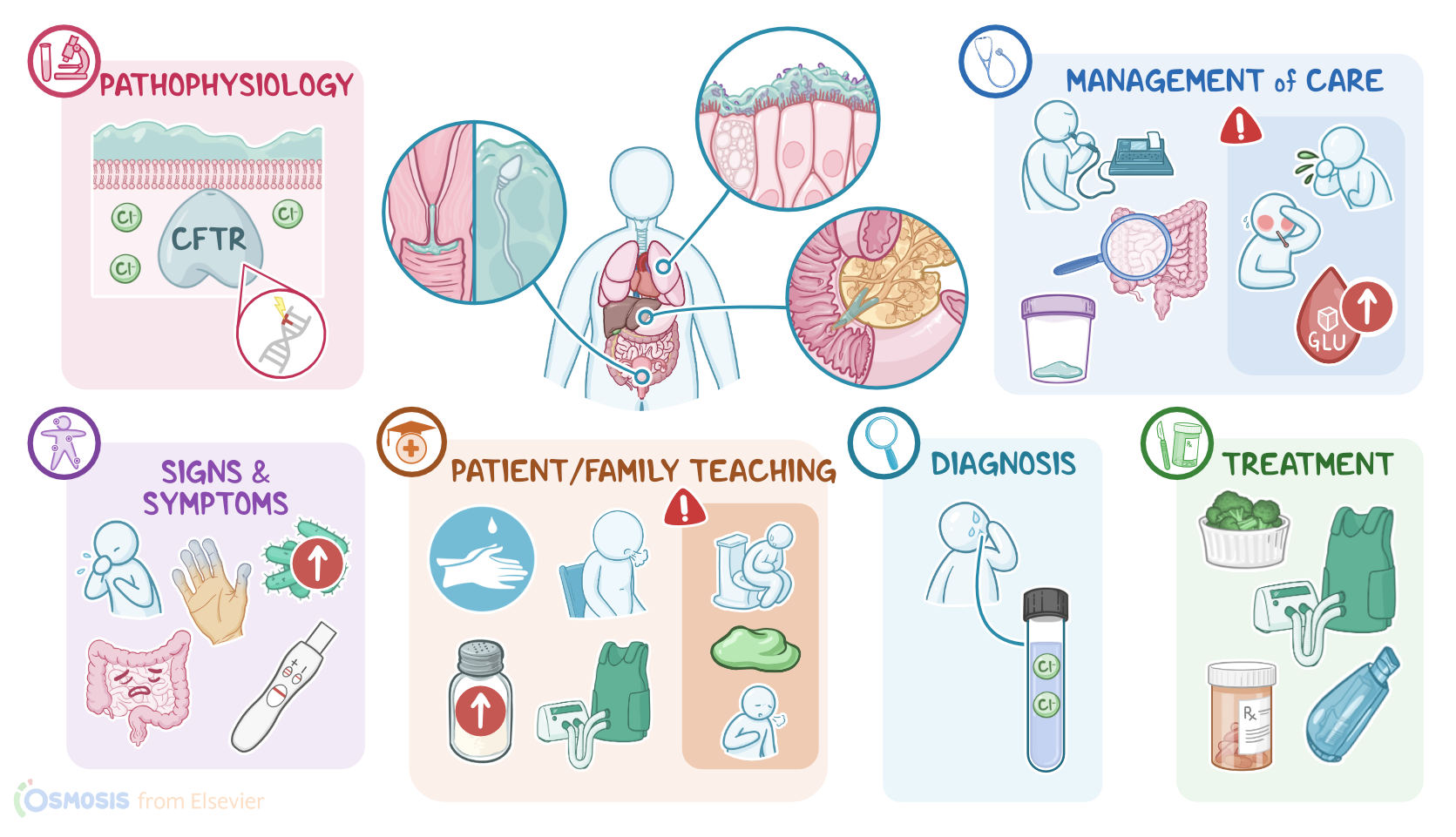
Transcript
Cystic fibrosis is an inherited, chronic and progressive condition characterized by dysfunction of the exocrine glands, primarily in the respiratory, gastrointestinal, and reproductive systems.
First, let’s quickly review some physiology. Exocrine glands are found throughout the body, and their main function is to secrete products like sweat from the skin; as well as saliva, gastric or pancreatic juices, and bile from the gastrointestinal tract; in addition to mucus to protect the respiratory, gastrointestinal, and reproductive systems, as well as seminal fluid from the prostate.
Now, zooming in, the exocrine glands consist of cells that secrete their products containing substances like water, ions, and enzymes into a ductal system. To do so, these cells have a special transporter protein called cystic fibrosis transmembrane conductance regulator protein, or CFTR for short. This protein pumps chloride ions out of the cell in order to help draw water out, forming those products and thinning them out, as needed for secretion.
Alright, so the main cause of cystic fibrosis is a mutation in the CFTR gene, which codes for the CFTR protein. This mutation is inherited with an autosomal recessive pattern, meaning that an individual needs to inherit one mutated gene from each parent to develop the condition. Thus, the main risk factor for cystic fibrosis is family history. In addition, another risk factor seems to be race and ethnicity, being more prevalent among individuals of white race.
Now, the pathology of cystic fibrosis develops because the mutated CFTR protein can’t insert into the cell membrane. As a result, exocrine cells can’t pump chloride ions out, so water doesn’t get drawn in, and the secretions are left overly thick. This can affect various organs in different ways.
In the respiratory tract, the thick mucus traps pathogens but can’t be swept up and out by cilia. As a consequence, these pathogens are able to remain in the lungs and keep replicating, which can lead to chronic respiratory infections, like pneumonia, and complications such as bronchiectasis, and even pneumothorax.
In the gastrointestinal tract, thick secretions can plug the pancreatic ducts, preventing the secretion of digestive enzymes into the small intestine. Over time, the backed-up digestive enzymes can degrade the cells lining the pancreatic ducts, which causes local inflammation and damage. This can ultimately lead to complications like pancreatitis, pancreatic insufficiency, and even diabetes.
In addition, the lack of digestive enzymes in the small intestine can result in complications like indigestion, malabsorption and eventually malnutrition, as well as thickened stools, which may lead to intestinal obstruction. The thick secretions can also plug the bile ducts, leading to the build up of bile that can damage the biliary tract, gallbladder, and even the liver. This process can cause complications like gallstones and, over time, biliary cirrhosis.
Now, in the male reproductive system, seminal fluid is abnormally thick, so sperm transport is impaired, causing complications like infertility. On the other hand, in the female reproductive system, cystic fibrosis can lead to abnormally thick cervical mucus, which makes it harder for spermatozoa to successfully pass through the cervix; this could cause subfertility, meaning pregnancy is difficult to achieve.
Additionally, in the skin, the excess chloride cannot be absorbed, so it remains trapped in sweat and attracts water, causing increased sweating and potentially dehydration; and also attracts sodium ions, which crystallize with chloride ions to form salt.
Finally, in the skeletal system, a combination of factors, including frequent infections, poor absorption of calcium and fat-soluble vitamins like vitamin D and K, and reproductive abnormalities, can impair bone development, causing lower bone mineral density and osteoporosis.
The clinical manifestations of cystic fibrosis primarily involve the respiratory tract. So, clients typically present with cough, wheezing, and cyanosis, which over time, can manifest as clubbing of fingers. In addition, these clients will be more susceptible to respiratory infections.
Next, clients with cystic fibrosis can also present with gastrointestinal symptoms. These typically include gastroesophageal reflux disease, as well as steatorrhea, and malnutrition, which can ultimately cause failure to thrive. In addition, clients can develop intestinal obstruction, which causes nausea, vomiting, and abdominal pain. An important sign in newborns is meconium ileus, which is a surgical emergency where the newborn’s first stools can get so thick and sticky that it might get stuck in the baby’s intestines and not come out. Moving on, cystic fibrosis can also cause reproductive symptoms. These include infertility or subfertility, as well as irregular menstruation and delayed puberty.
Additionally, clients may experience excessive sweating, which can lead to dehydration. In addition, affected clients may have a salty skin, which is often reported by parents when kissing their child.
The diagnosis of cystic fibrosis starts with the client's history and physical assessment. The gold standard test to diagnose cystic fibrosis is a sweat chloride test, which helps detect high amounts of chloride ions in the sweat of a client with cystic fibrosis. Other diagnostic tests include genetic testing, which can be done to confirm the diagnosis; as well as newborn screening tests, which can aid in early detection of the condition.