Dravet Syndrome · What It Is, Causes, Treatment, and More
Published: Feb 04, 2025
Author: Emily Miao, PharmD•
Editor: Alyssa Haag•
Editor: Józia McGowan, DO•
Editor: Kelsey LaFayette, DNP
Illustrator: Abbey Richard
7-day free trial
Go deeper with Osmosis
Osmosis is a learning platform with videos, questions, and AI tools to help you master topics like this.
Watch quick, visual videos
Practice with Qbank-style questions
Use AI to explain, quiz, and review
Study anytime with the mobile app
No credit card · Cancel anytime
What is Dravet syndrome?
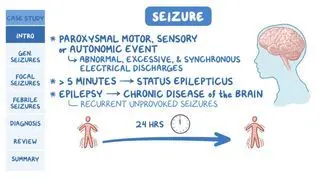
Dravet syndrome, previously known as severe myoclonic epilepsy of infancy, is a rare genetic syndrome characterized by recurrent seizures and refractory epileptogenic encephalopathies (i.e., abnormal brain activity that is not responsive to anti-seizure medications), resulting in long-term neurocognitive sequelae. It is a rare condition, occurring in an estimated 1 in 15,700 to 40,000 live births, affects all genders equally, and commonly presents in childhood. A mutation in the SCN1A gene has been identified in over 70% of individuals with Dravet syndrome. Children with Dravet syndrome typically experience their first seizure in their infantile years of life and experience neurodevelopmental decline, leading to premature mortality. Individuals may experience many forms of epilepsy and seizure types throughout their lifetime, including absence seizures, tonic-clonic seizures, and (rarely) status epilepticus.
Learn deeper with Osmosis
Master this topic faster with videos, questions, and AI.
Used by 8M+ healthcare learners.
Start free trial
No credit card · Cancel anytime
What causes Dravet syndrome?
Dravet syndrome is caused by a mutation in the SCN1A gene, which codes for a voltage-gated sodium channel located primarily in neuronal cell bodies and dendrites. It is therefore hypothesized that impairment of these channels leads to loss of proper neuronal firing, thereby impairing the body’s neural circuits. Approximately 90% of SCN1A gene mutations arise de novo, meaning not inherited from one’s parents. In 5-10% of cases, the SCN1A mutation is passed on to a child from either parent. The risk factors for Dravet syndrome are poorly understood; however, the most common seizure-provoking precipitants reported by caregivers include fever, illness, physical activity, and recent immunization.
What are the signs and symptoms of Dravet syndrome?
Signs and symptoms of Dravet syndrome include a combination of various forms of epilepsy, neurodevelopmental delay, and neurologic disability that begin after the onset of seizures. Those with Dravet syndrome are typically previously healthy and experience their first seizure within their first year of life. The first seizure is usually a tonic-clonic seizure (i.e., grand mal), which involves rhythmic stiffening and violent jerking of the limbs along with loss of consciousness. This type of seizure may involve one or both sides of the body and be associated with tongue-biting, urinary incontinence, and/or defecation. Individuals may also be confused or lethargic following the seizure, known as a postictal state. Rarely, the seizure may evolve to status epilepticus, a medical emergency consisting of a seizure lasting longer than 5 minutes or multiple seizures occurring within 5 minutes.
Following the first seizure, individuals may also experience absence seizures and/or recurrent focal or generalized tonic-clonic epilepsy. Absence seizures (i.e., petit mal) are more common in children and are characterized by blankly staring into space, usually lasting for a few seconds.
Individuals with Dravet syndrome may also have neurodevelopmental delays and present with generalized hypotonia (i.e., decreased muscle tone), ataxia (i.e., impaired balance and coordination), and dysautonomia (i.e., inability to regulate the autonomic nervous system). With dysautonomia, individuals may be unable to regulate their body temperature or heart rate, which may lead to overheating and, consequently, increased frequency of seizures. These developmental delays begin shortly after the first seizure and continue to worsen over time. Individuals may also have comorbid behavioral disturbances and conditions, including autism spectrum disorder, aggression, irritability, and maladaptive psychosocial behaviors.
How is Dravet syndrome diagnosed?
The diagnosis of Dravet syndrome is clinical and begins with a thorough history and review of symptoms following the first seizure. Several clinical features that aid the diagnosis include the age of onset, seizure patterns and triggers, and neurodevelopmental delays. Initial evaluation includes a neurologic examination, laboratory blood testing, and genetic testing for the SCN1A mutation. Genetic testing for SCN1A mutation may be negative in up to 20% of individuals; however, the diagnosis cannot be excluded.
Further imaging, such as magnetic resonance imaging (MRI) and electroencephalography (EEG), are not required for diagnosis but may be helpful to exclude other differential diagnoses, such as an intracranial neoplasm causing recurrent seizure activity. People with Dravet syndrome will often have normal MRI findings and evolving EEG findings depending on the type of seizures experienced. For example, the EEG is usually normal during the initial seizure, but EEG findings between seizures (i.e., interictal) range from generalized spike waves to isolated brief bursts. An EEG may also be helpful to confirm the complete cessation of an epileptogenic episode and to rule out status epilepticus.
Further imaging, such as magnetic resonance imaging (MRI) and electroencephalography (EEG), are not required for diagnosis but may be helpful to exclude other differential diagnoses, such as an intracranial neoplasm causing recurrent seizure activity. People with Dravet syndrome will often have normal MRI findings and evolving EEG findings depending on the type of seizures experienced. For example, the EEG is usually normal during the initial seizure, but EEG findings between seizures (i.e., interictal) range from generalized spike waves to isolated brief bursts. An EEG may also be helpful to confirm the complete cessation of an epileptogenic episode and to rule out status epilepticus.
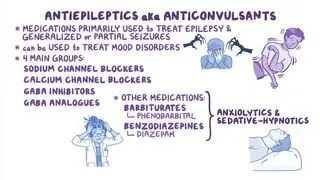
How is Dravet syndrome treated?
Dravet syndrome is treated with a combination of pharmacologic and nonpharmacologic modalities aimed at reducing both the length and number of seizures, preventing status epilepticus, and improving the individual’s quality of life. The first-line anticonvulsant medication used to treat seizures in this population is valproate, a sodium channel blocker that increases levels of gamma-aminobutyric acid (GABA), a neurotransmitter that helps maintain the inhibitory tone in the brain. In those who do not respond to initial treatment, other adjuncts can include clobazam, stiripentol, fenfluramine, topiramate, cannabidiol, and vagus nerve stimulation. Several sodium-blocking anticonvulsants, including carbamazepine, oxcarbazepine, lamotrigine, and phenytoin, have been reported to aggravate seizures and are typically avoided.
As individuals with Dravet syndrome tend to be more sensitive to certain seizure triggers compared to the general population with epilepsy, avoidance of triggers (e.g., fever, hyperthermia, and vaccinations) is an important aspect of management. Fever and hyperthermia can be managed by avoiding hot baths or high-impact physical activity on warm days. Oral and rectal antipyretics, including acetaminophen and ibuprofen, can be used in conjunction to lower the body’s temperature in the setting of febrile seizures. While various vaccinations may be seizure triggers, the highest risk is observed in the measles, mumps, and rubella (MMR) vaccine. Nonetheless, all age-appropriate vaccinations can still be offered based on the individual's benefits and the risks. After vaccinations, it may be important to monitor individuals with Dravet syndrome for post-vaccination seizures.
Finally, it is important to devise and distribute an individualized emergency action plan to family, friends, and caregivers. This plan usually includes a benzodiazepine for emergency treatment and instructions in the event the seizure progresses to status epilepticus.
What are the most important facts to know about Dravet syndrome?
Dravet syndrome, previously known as severe myoclonic epilepsy of infancy, is a rare genetic syndrome characterized by recurrent seizures and refractory epileptogenic encephalopathies, resulting in long-term neurocognitive sequelae. It is caused by a mutation in the SCN1A gene, which codes for a voltage-gated sodium channel. Signs and symptoms include a combination of various forms of epilepsy, neurodevelopmental delay, and neurologic disability; which begins after seizure onset. While the diagnosis is clinical, several features that may aid the clinician in the diagnosis include the age of onset, seizure patterns and triggers, and neurodevelopmental delays following the onset of seizures. Treatment is aimed at reducing both the length and number of seizures, preventing status epilepticus, and improving the individual’s quality of life.
Related topics


Students say Osmosis is 100% worth it
Because Osmosis saves them time. Lowers stress. And actually helps them remember when it counts.
I used Osmosis to prepare for my first medical school licensing exam! Super helpful and interactive for people who may not do great with just pages of text info!
Cecilia Ruiz
MD student

I have used Osmosis for about four years. Best thing I have ever used for my medical studies.
Sayan Misra
Med student
Osmosis videos are superior because they define simple concepts, tell a story with a clear progression, and provide context.
Jay Pate
Dental student
References
Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135(8):2329-36. doi: 10.1093/brain/aws151.
He Z, Li Y, Zhao X, Li B. Dravet syndrome: Advances in etiology, clinical presentation, and treatment. Epilepsy Res. 2022;188:107041. doi: 10.1016/j.eplepsyres.2022.107041.
Skluzacek JV, Watts KP, Parsy O, Wical B, Camfield P. Dravet syndrome and parent associations: The IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia. 2011;52(Suppl 2):95-101. doi: 10.1111/j.1528-1167.2011.03012.x.
Wheless JW, Fulton SP, Mudigoudar BD. Dravet syndrome: A review of current management. Pediatr Neurol. 2020;107:28-40. doi: 10.1016/j.pediatrneurol.2020.01.005.