Myasthenia gravis: Nursing
Myasthenia gravis: Nursing
musculoskeletal
musculoskeletal
Notes
| MYASTHENIA GRAVIS | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
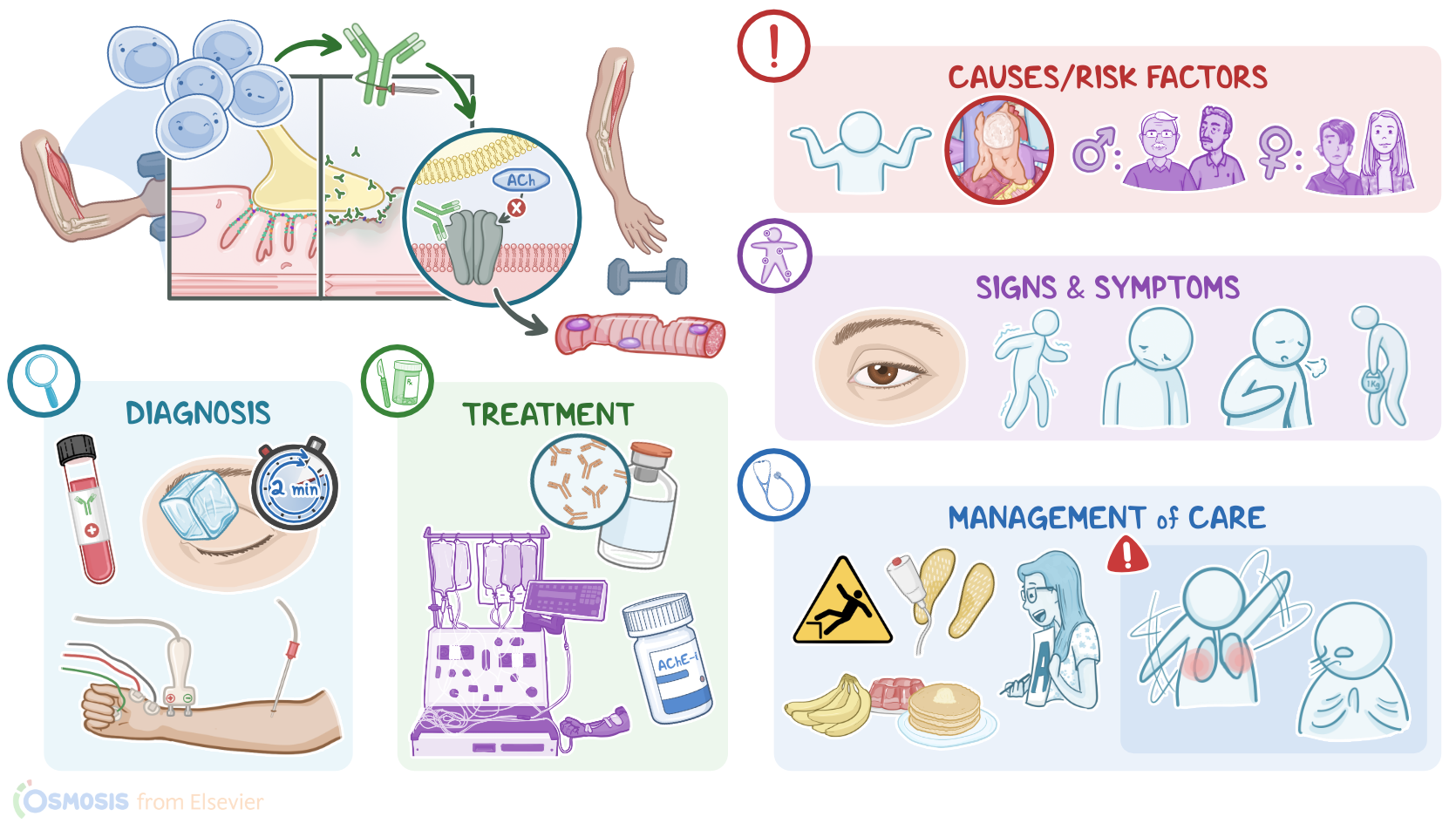
Transcript
Myasthenia gravis is a chronic, progressive autoimmune disease caused by antibodies that bind to and destroy neuromuscular communication, where nerves send impulses to trigger skeletal muscle contraction. Over time, this results in fatigue and muscle weakness due to impaired muscle contractions.
Now, to better understand myasthenia gravis, let's review the normal physiological process of muscle contraction that occurs at cellular level. Muscle contraction is initiated at the neuromuscular junction, which is basically a connection between the axonal end of a motor neuron and a skeletal muscle fiber. So, the end of the neuron is called the presynaptic membrane, while the muscle cell is called the postsynaptic membrane.
Now, to trigger muscle contraction, the axonal end of the motor neuron releases the neurotransmitter acetylcholine, also called ACh, at the neuromuscular junction. Acetylcholine then binds to the nicotinic acetylcholine receptors present on the muscle cell membrane. The binding of acetylcholine to its receptor activates a chain reaction within the muscle cell, which ultimately results in voluntary muscle contraction.
Now, myasthenia gravis is caused by autoantibodies that target the neuromuscular junction. In a minority of cases, this occurs as a paraneoplastic syndrome, where an underlying cancer like thymoma or bronchogenic carcinoma can result in the production of autoantibodies. However, in the majority of cases, myasthenia gravis does not have a clear underlying cause. Clients at risk are those who are assigned female at birth in their 20s and 30s, or clients assigned male at birth in their 60s and 70s.
So, in myasthenia gravis, the client's own B cells generate autoantibodies, setting in a type II hypersensitivity reaction. These autoantibodies specifically bind with the postsynaptic acetylcholine receptor, or AChR for short, in 90% of clients, or sometimes target the muscle-specific tyrosine kinase receptors, or MuSK for short, in 10% of clients, both of which ultimately lead to loss of acetylcholine receptor function. In addition, binding of autoantibodies to acetylcholine receptors activates the classical pathway of the complement, which triggers inflammation and damage of the postsynaptic membrane of muscle cells, ultimately decreasing the number of available acetylcholine receptors on the muscle membrane. As a result of all these, initiation of a muscle contraction becomes less effective over time.
Typically, myasthenia gravis presents with fatigue and skeletal muscle weakness that fluctuates throughout the day. Clients with myasthenia gravis might wake up feeling fine, but get progressively weaker as their daily activities go on, and by the end of the day they might feel very weak. This weakness usually increases when performing repetitive movements, and improves with rest. Moreover, myasthenia gravis is a chronic and progressive disease that worsens over time.
The initial symptoms of myasthenia gravis often involve the extraocular muscles, which control the movement of the eyes and eyelids, so clients might experience diplopia or ptosis. If facial muscles are involved, clients may present with impaired facial expressions, difficulty in chewing or swallowing, slurred speech, and fading out voice along the conversation. Neck muscles can become weak too, and these clients might feel their head is heavy and it’s hard to keep upright. As the disease progresses, arm and leg muscles are also involved, so clients might experience difficulty with walking, climbing stairs, and even performing simple tasks such as hair combing and brushing their teeth. Clients may also develop an abnormal posture due to weakness of the hip muscles.
Myasthenia gravis can also affect the respiratory muscles, so the diaphragm and the intercostal muscles, leading to difficulty breathing. Finally, a potentially life-threatening acute exacerbation of the disease is a myasthenic crisis, which can present with a worsening generalized muscle weakness, and even lead to respiratory failure. Triggers of myasthenic crisis include stressful events like an infection, surgery, emotional trauma, temperature extremes, or pregnancy. Lastly, certain medications can make myasthenia gravis worse, and should be avoided or used with caution, including certain antibiotics like fluoroquinolones, macrolides, and aminoglycosides; as well as beta-blockers like propranolol; and antiarrhythmic medications like procainamide and quinidine.
Now, the diagnosis of myasthenia gravis starts with a history and physical assessment, as well as laboratory tests to detect AChR or MuSK antibodies in blood. Additional diagnostic tests include electromyography, or EMG for short, which uses repetitive electrical stimuli to assess muscle contraction. A Tensilon test can be performed by intravenously injecting edrophonium chloride, which is an acetylcholinesterase inhibitor that briefly blocks the breakdown of acetylcholine and temporarily increases its levels at the neuromuscular junction; in myasthenia gravis, this is enough to overcome the symptoms caused by the decreased number of available receptors, so the weakness is improved. Lastly, chest X-ray and CT scan can be performed to rule out an underlying cause like a thymoma.
Although there’s no cure for myasthenia gravis, certain medications can be used to help mitigate some of the symptoms and improve the client’s quality of life. Initial treatment consists of acetylcholinesterase or AChE inhibitors, such as neostigmine or pyridostigmine. These medications prevent the enzyme acetylcholinesterase from degrading acetylcholine, which increases its concentration around muscle cells and, subsequently, its effects are increased and prolonged. Severe cases of myasthenia gravis can also be treated with immunosuppressive medications, such as corticosteroids like prednisone, which reduce the production of autoantibodies.
Clients experiencing an acute exacerbation or myasthenic crisis can be treated with intravenous immune globulins or IVIGs, in order to quickly neutralize the autoantibodies and stop the acute exacerbation; or with plasmapheresis, or plasma exchange to filter out the circulating autoantibodies from the client’s blood; and some clients may require respiratory support. Finally, clients with a thymoma can undergo surgical removal of the thymus, known as thymectomy.
All right, now let’s look at nursing care for a client with myasthenia gravis. The main goals of nursing care are to prevent and manage complications, as well as promote optimal muscle strength and improved quality of life. Begin assessing your client’s muscle weakness, including the onset, duration, and frequency of muscle weakness. Assess your client’s respiratory rate, depth, effort, breathing pattern, breath sounds, and oxygen saturation.
Report concerning assessment findings, like a respiratory rate below 12 or above 20 breaths per minute, an oxygen saturation below 92%, abnormal chest movement, or reduced or absent breath sounds. If this occurs, ensure oxygen, suction equipment, and a bag valve mask are readily available. Be sure to also assess your client’s ability to cough and swallow, and implement aspiration precautions.