Cystic fibrosis: Nursing
Introduction0:00–0:17
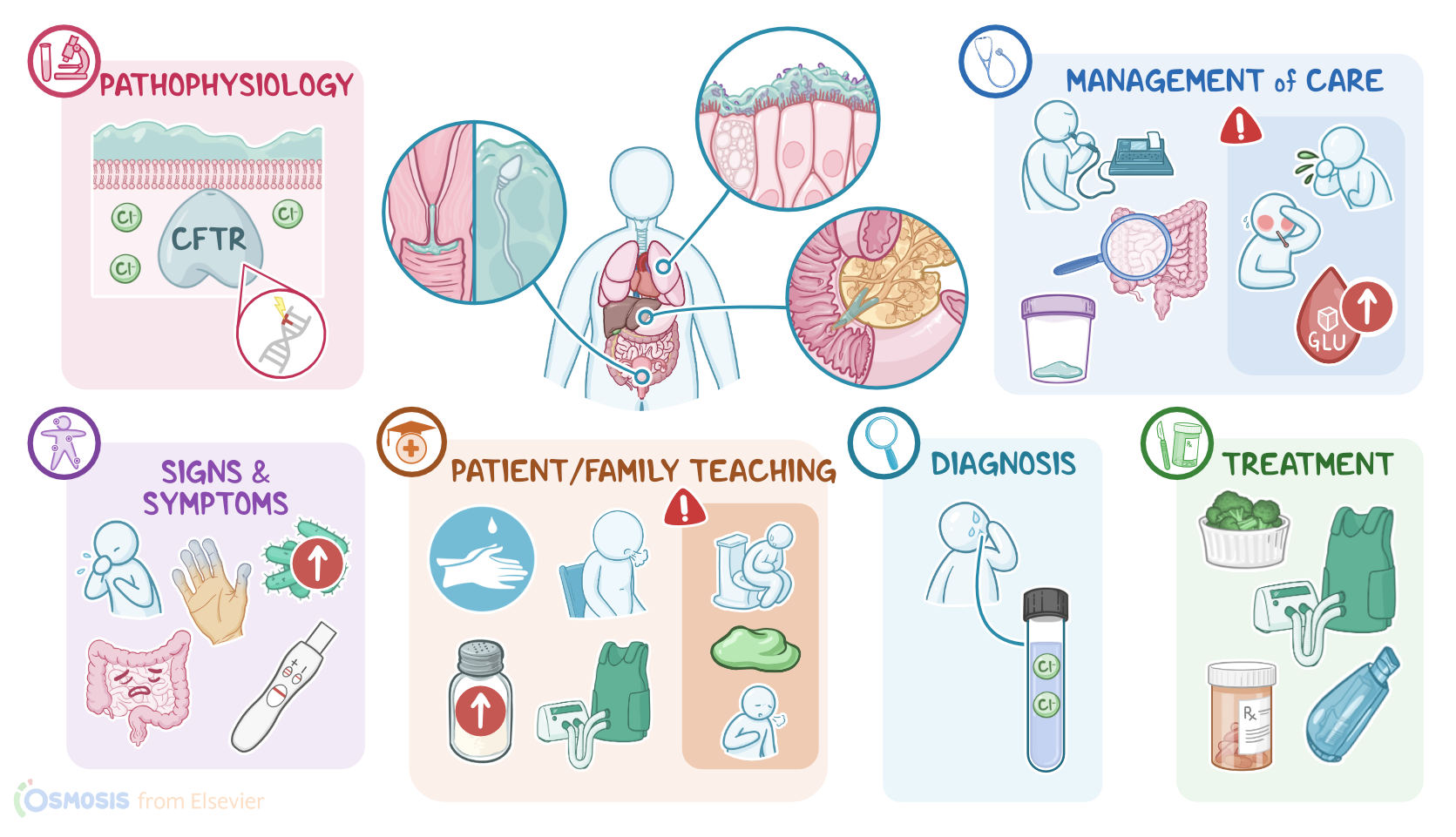
Cystic fibrosis is an inherited, chronic and progressive condition characterized by dysfunction of the exocrine glands, primarily in the respiratory, gastrointestinal, and reproductive systems.
Physiology0:17–1:16
First, let’s quickly review some physiology. Exocrine glands are found throughout the body, and their main function is to secrete products like sweat from the skin; as well as saliva, gastric or pancreatic juices, and bile from the gastrointestinal tract; in addition to mucus to protect the respiratory, gastrointestinal, and reproductive systems, as well as seminal fluid from the prostate.
Now, zooming in, the exocrine glands consist of cells that secrete their products containing substances like water, ions, and enzymes into a ductal system.
To do so, these cells have a special transporter protein called cystic fibrosis transmembrane conductance regulator protein, or CFTR for short.
This protein pumps chloride ions out of the cell in order to help draw water out, forming those products and thinning them out, as needed for secretion.Alright, so the main cause of cystic fibrosis is a mutation in the CFTR gene, which codes for the CFTR protein.
Causes & risk factors1:16–1:50
This mutation is inherited with an autosomal recessive pattern, meaning that an individual needs to inherit one mutated gene from each parent to develop the condition.
Thus, the main risk factor for cystic fibrosis is family history. In addition, another risk factor seems to be race and ethnicity, being more prevalent among individuals of white race.
Pathology1:50–4:42
Now, the pathology of cystic fibrosis develops because the mutated CFTR protein can’t insert into the cell membrane. As a result, exocrine cells can’t pump chloride ions out, so water doesn’t get drawn in, and the secretions are left overly thick.
This can affect various organs in different ways. In the respiratory tract, the thick mucus traps pathogens but can’t be swept up and out by cilia.
As a consequence, these pathogens are able to remain in the lungs and keep replicating, which can lead to chronic respiratory infections, like pneumonia, and complications such as bronchiectasis, and even pneumothorax.
In the gastrointestinal tract, thick secretions can plug the pancreatic ducts, preventing the secretion of digestive enzymes into the small intestine.
Over time, the backed-up digestive enzymes can degrade the cells lining the pancreatic ducts, which causes local inflammation and damage.
This can ultimately lead to complications like pancreatitis, pancreatic insufficiency, and even diabetes. In addition, the lack of digestive enzymes in the small intestine can result in complications like indigestion, malabsorption and eventually malnutrition, as well as thickened stools, which may lead to intestinal obstruction.
The thick secretions can also plug the bile ducts, leading to the build up of bile that can damage the biliary tract, gallbladder, and even the liver.
This process can cause complications like gallstones and, over time, biliary cirrhosis.Now, in the male reproductive system, seminal fluid is abnormally thick, so sperm transport is impaired, causing complications like infertility.
On the other hand, in the female reproductive system, cystic fibrosis can lead to abnormally thick cervical mucus, which makes it harder for spermatozoa to successfully pass through the cervix; this could cause subfertility, meaning pregnancy is difficult to achieve.
Additionally, in the skin, the excess chloride cannot be absorbed, so it remains trapped in sweat and attracts water, causing increased sweating and potentially dehydration; and also attracts sodium ions, which crystallize with chloride ions to form salt.
Finally, in the skeletal system, a combination of factors, including frequent infections, poor absorption of calcium and fat-soluble vitamins like vitamin D and K, and reproductive abnormalities, can impair bone development, causing lower bone mineral density and osteoporosis.
The clinical manifestations of cystic fibrosis primarily involve the respiratory tract. So, clients typically present with cough, wheezing, and cyanosis, which over time, can manifest as clubbing of fingers.
Clinical manifestations4:42–6:04
In addition, these clients will be more susceptible to respiratory infections. Next, clients with cystic fibrosis can also present with gastrointestinal symptoms.
These typically include gastroesophageal reflux disease, as well as steatorrhea, and malnutrition, which can ultimately cause failure to thrive.
In addition, clients can develop intestinal obstruction, which causes nausea, vomiting, and abdominal pain. An important sign in newborns is meconium ileus, which is a surgical emergency where the newborn’s first stools can get so thick and sticky that it might get stuck in the baby’s intestines and not come out.
Moving on, cystic fibrosis can also cause reproductive symptoms. These include infertility or subfertility, as well as irregular menstruation and delayed puberty.
Additionally, clients may experience excessive sweating, which can lead to dehydration. In addition, affected clients may have a salty skin, which is often reported by parents when kissing their child.
The diagnosis of cystic fibrosis starts with the client's history and physical assessment. The gold standard test to diagnose cystic fibrosis is a sweat chloride test, which helps detect high amounts of chloride ions in the sweat of a client with cystic fibrosis.
Diagnosis6:04–6:30
Other diagnostic tests include genetic testing, which can be done to confirm the diagnosis; as well as newborn screening tests, which can aid in early detection of the condition.
The treatment of cystic fibrosis is typically geared at easing up the symptoms and preventing complications. Clients with gastrointestinal symptoms can be treated with pancreatic enzyme replacement, as well as vitamin supplementation, and improved nutrition to promote healthy weight gain.
Treatment6:30–7:58
On the other hand, respiratory symptoms can be treated with supplemental oxygen; as well as chest physiotherapy, which includes breathing exercises, percussion, vibration, and postural drainage.
Other options are positive expiratory devices to promote the elimination of mucus. These include oscillating positive pressure devices that create vibrations in the large and small airways when the client breathes into them, as well as a vest that can be worn that vibrates the chest in order to loosen mucus in the airways.
Additionally, clients with respiratory symptoms can be treated with medications, such as bronchodilators, as well as mucolytic medications that reduce the mucus thickness, and antibiotics for respiratory infections.
Another therapeutic option in clients with certain CFTR gene mutations is using a CFTR modulator, like lumacaftor or ivacaftor.
These medications improve the function of the impaired CFTR protein. Finally, clients with severe loss of pulmonary function over time can be treated surgically with lung transplantation, which can make a substantial difference in their daily life.Alright, let’s talk about the nursing care you’ll provide for a client with cystic fibrosis.
Management and care7:58–10:47
Priority goals are to promote airway clearance and gas exchange, monitor for infection, and monitoring for pulmonary and non-pulmonary complications of cystic fibrosis.
Begin by assessing your client’s vital signs, with a focus on their respiratory status. Check for the presence of chest congestion, cough, and sputum production.
Then assess their oxygen saturation, rate and depth of breathing, breath sounds, and use of accessory muscles. Review results of their pulmonary function tests, including their forced vital capacity, or FVC, and forced expiratory volume, or FEV1 tests.
Report assessment findings that indicate a pulmonary exacerbation is occurring like decreased SpO2, productive cough, new or worsening crackles heard on auscultation, intercostal retractions, or a 10% or more reduction in FEV1.
Administer supplemental oxygen and prescribed medications, including bronchodilators and mucolytics; and assist the respiratory therapist in performing chest physiotherapy.To prevent infection, assign your client to a private room if possible and have them wear a surgical mask in all common areas.
Then monitor them for signs of lung infection by regularly assessing their temperature and evaluating them for cough and sputum production.
Collect a complete blood count, or CBC, and sputum sample. Report fever of 100.4 F or 38.0 C, elevated white blood cell count, or a positive sputum culture to the healthcare provider.
Then administer the prescribed antibiotics. Next, let’s talk about non-pulmonary complications of cystic fibrosis.
Assess for signs of pancreatic enzyme insufficiency and impaired nutrition, including steatorrhea, unintended weight loss, and delayed growth in children.
Monitor for signs of osteoporosis and monitor bone growth in children, and ensure your client is eating a diet high in calories, protein, iron, salt and calcium.
Administer pancreatic enzyme supplements and multivitamin as prescribed, and collaborate with the dietitian to support your client’s nutritional needs.
Then, review their laboratory test results, and report a postprandial blood glucose level of more than 200 mg/dL or a fasting glucose greater than 130 mg/dL.
General client and family teaching10:47–13:56
Alright, now let’s move on to teaching. Begin by explaining that cystic fibrosis is a genetic disorder that makes secretions in the body thicker, which causes problems in the lungs, pancreas, liver, intestines, and reproductive systems.
Then, remind them that thick mucus in their lungs increases their risk of developing serious infections, and review infection control measures they need to incorporate in their daily routine, including practicing good hand hygiene, disinfecting surfaces regularly, avoiding large crowds, and keeping up to date with their immunizations.
Lastly, stress the importance of maintaining a smoke free environment.Next, stress the importance of adhering to their treatment plan to reduce the frequency of exacerbations, and teach them how to self-administer their medications as directed.
Then, review airway clearance techniques. For huff coughing, instruct them to sit up straight and takes a slow, deep breath, and hold it for 2-3 seconds; then use their diaphragm and stomach muscles to make a series of rapid exhalations, making a “ha” sound with each exhalation, as if they were trying to fog a mirror with their breath.
Have them repeat this technique 2-3 times. Lastly, collaborate with the respiratory therapist to review with caregivers technique for chest physiotherapy, including chest percussion and postural drainage.
If your client is prescribed an oscillating vest or oscillating positive pressure device, collaborate with the respiratory therapist to provide hands on training for proper use and maintenence of these devices.
Also talk to them about how thick secretions in their pancreas and liver can cause problems with nutrition, and discuss how consuming extra calories and nutrients can support growth and development, promote healthy lungs, and to help them fight infection.
Remind them to eat a high calorie diet that includes protein rich foods like meat, eggs, soy, and beans; iron rich foods like fortified cereals and dark green vegetables; and calcium rich foods like full fat dairy products.
Also remind them to take a daily multivitamin that contains the vitamins A, D, E, and K; and teach them to increase their salt intake during hot weather or when they are sweating more during exercise.
Lastly, remind them about the importance of physical activity in promoting overall health and airway clearance, and collaborate with the physical therapist to design an individualized exercise plan.Finally, instruct them to contact the healthcare provider right away if they experience symptoms like severe constipation, diarrhea or abdominal pain; if they are producing more mucus than usual or if the mucus is dark, or greenish in color; or if they experience a sudden shortness of breath; if they are more tired than usual, or if they develop a fever.Alright, as a quick recap… Cystic fibrosis is an inherited disorder that causes a defect in the CFTR protein, a protein involved in chloride ion transport.
Review13:56–15:40
As a result, mucus becomes very thick, leading to complications throughout the body.Manifestations are primarily in the respiratory system and include cough, wheezing, cyanosis, clubbing of the fingers, and frequent respiratory infections.
In the gastrointestinal tract, manifestations include gastroesophageal reflux, malabsorption, steatorrhea, bowel obstruction, and gallstone development.
Reproductive manifestations include infertility, irregular menses, and delayed onset of puberty, and integumentary manifestations include salty skin, while excessive sweating can lead to dehydration.
Genetic testing can be done, and newborn screening tests can provide early identification of the disorder. Treatment is centered around controlling symptoms and preventing complications, and involves medications such as bronchodilators,
| CYSTIC FIBROSIS | ||
| KEY POINTS | NOTES | |
| DEFINITION |
| |
| PHYSIOLOGY |
| |
| CAUSES AND RISK FACTORS |
| |
| PATHOPHYSIOLOGY |
| |
| SIGNS AND SYMPTOMS |
| |
| DIAGNOSIS |
| |
| TREATMENT |
| |
| MANAGEMENT OF CARE |
| |
| PATIENT AND FAMILY TEACHING |
| |
No notes for this video yet
Try adding a note below